 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of the tryptophan–terbium FRET pair coupled to silver nanoprisms for biosensing applications
Ane K.
di Gennaro
a,
Leonid
Gurevich
a,
Esben
Skovsen
a,
Michael T.
Overgaard
b and
Peter
Fojan
*a
aDepartment of Physics and Nanotechnology, Aalborg University, Skjernvej 4A, 9220 Aalborg, Denmark. E-mail: fp@nano.aau.dk; Fax: +45 99409235; Tel: +45 99407488
bDepartment of Biotechnology, Chemistry and Environmental Engineering, Aalborg University, Sohngaardsholmsvej 47, 9000 Aalborg, Denmark. E-mail: mto@bio.aau.dk; Tel: +45 99408525
First published on 16th April 2013
Abstract
Plasmonic coupling between fluorophores and metal surfaces has become a focal point of optical research during the last two decades, however, the interactions of FRET couples with metal surfaces remain relatively unexplored. In this study, interactions of the tryptophan–Tb3+ FRET pair with silver nanoprisms for potential biosensor development have been investigated. For this purpose an engineered lanthanide binding peptide (LBTtrp) containing tryptophan as the sensitizer for bound lanthanide ions (Tb3+) as well as a trypsin cleavage site was synthesized. The modified LBTtrp peptide contained two N-terminal cysteine residues to provide a stronger coupling to the silver nanoprisms (∼6 nm high, ∼50 nm wide). This study investigated the interaction between tryptophan, chelated Tb3+ ions, and silver nanoprisms in solution using fluorescence and transient absorption spectroscopy. We have found that Tb3+ luminescence decreases upon binding of the LBTtrp–Tb3+ to silver nanoprisms and increases upon trypsin cleavage. The transient absorption spectroscopy measurements showed a significant decrease in the lifetime of the excited singlet state of tryptophan upon Tb3+ chelation, while coupling to the silver nanoprisms did not show a significant effect on tryptophan. The results obtained in this work demonstrate a first proof of concept for a new sensitive optical biosensor in solution.
Introduction
Nanomaterials science has evolved from dealing with pure physics and inorganic materials towards the development of advanced building blocks for biotechnological applications. Their unique properties have the potential to boost the development of new biosensors and to drastically broaden their application within environmental, industrial and biomedical sensing applications. This also includes medical diagnostics, proteomics and drug discovery. The increasing demand for enhanced sensitivity as well as improved selectivity of the biosensors spurs optimization and development of new types of biosensors. The phenomenon of localized plasmon resonance (LPR) in metallic nanoparticles has made them one of the main subjects of interest for the development of new biosensors.1,2 As a remarkable example, strong LPR scattering of gold nanoparticles coupled to fluorescently labeled biomolecules has improved the diagnostics of cancer as well as several other diseases.1 Recent developments in nanoparticle synthesis technology allow for the efficient production of metal nanoparticles with tunable absorption spectra (colors).3 This can be achieved by varying the shape and dimensions of the metal nanoparticle4 or the metal shell in a core–shell particle5 and thus, changing the metal interaction with fluorophores on the surface. Thin metal surfaces made of gold or silver are also known to induce quenching of excited states via energy transfer to the metal surface, which is a phenomenon of considerable interest in biophotonics.3,6For several years lanthanides have been used for protein labeling allowing for sensing and visualization of dynamical processes in proteins, localization of proteins in cells and tissues, as well as protein–protein interactions.7 For this purpose, peptide chelates for lanthanide ions have been designed and improved using protein engineering methods over the years. The chelate includes a lanthanide binding tag (LBT), comprising 15–20 amino acids and has to achieve several purposes: (1) high affinity and specific binding of the lanthanide ion, (2) provide the attachment of the lanthanide to a macromolecule, (3) prevent H2O coordination with the lanthanide, which otherwise would lead to luminescence quenching, and (4) contain a covalently coupled sensitizer molecule. The sensitizer molecule should exhibit a high absorption coefficient compared to the relatively small absorption cross section of lanthanides.8 A good sensitizer for such a system is tryptophan (Trp), which has a molar extinction coefficient of 5800 cm−1 M−1 at 280 nm, while the molar extinction coefficient of Tb3+ is approximately 0.2 cm−1 M−1 at the same excitation wavelength.9,10 Inserting such a sensitizer allows for lanthanide excitation using common fluorescence spectroscopy setups.8
Recent studies have shown that upon excitation of the sensitizer molecule, positioned in the LBT, the sensitizer transfers its energy to the lanthanide ion. This occurs via a three stage intersystem crossing mechanism involving transfer from the singlet excited state to the triplet state and then to the lanthanide ion (Fig. 1).11 The advantages of using lanthanides over molecular chromophores are: (1) long luminescence lifetimes, in the millisecond range, (2) narrow luminescence bands with large Stoke shifts (>200 nm), and (3) no bleaching.11,12 Long lifetimes offer the advantage of reducing the background fluorescence signal thus increasing the detection sensitivity.11
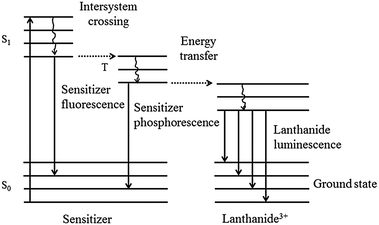 | ||
| Fig. 1 Energy diagram of the lanthanide complex, consisting of a sensitizer and a lanthanide(III) ion. Upon sensitizer excitation, intersystem crossing to the sensitizer triplet state leads to energy transfer to the lanthanide(III) ion. | ||
The photophysical and photochemical properties of Trp have been described in several reviews, discussing two different relaxation pathways from the singlet excited state of Trp (1Trp*). These two different pathways are described in eqn (1) and (2).
| Trp + hν → 1Trp* → 1Trp˙+ + e−aq | (1) |
| Trp + hν → 1Trp* → 3Trp | (2) |
The first relaxation pathway (eqn (1)) involves electron ejection from 1Trp* resulting in a positively charged Trp radical (1Trp˙+) and a solvated electron (e−aq). The second relaxation pathway (eqn (2)) involves intersystem crossing from the excited singlet state to the excited triplet state (3Trp).13,14
Already a few decades ago, effects related to plasmonic coupling of fluorophores such as enhanced fluorescence on colloidal metal surfaces were observed.15 Due to the increasing demand for high sensitivity biomarker detection, this interaction has been investigated intensively using different metal nanostructures such as gold or silver nanoparticles4 or silver island films.16,17 The distance between the fluorophore and the metal surface determines whether the fluorescence is enhanced or quenched. In order to enhance the fluorescence signal, the optimal surface–fluorophore distance should be around 10 nm, depending on the metal.18 When decreasing the distance, quenching by the metal dominates the interaction due to non-radiative energy transfer to the metal.17,18 On the other hand, the enhancement effect of the metal on the fluorescence emission decreases progressively with increasing separation distances between the fluorophore and the metal surface.15,19 In this respect, it can be expected that coupling and uncoupling of the tryptophan–Tb3+ complex from the metal surface would produce a significant effect on Tb3+ luminescence. To our knowledge the interaction of the Trp–Tb3+ FRET couple with a metal surface has not been reported before. The study of such an interaction could shed light on the energy transfer mechanism between 1Trp* and Tb3+ in the presence of silver nanoprisms (AgPr). From an applied science perspective, this effect could form the basis of an optical biosensor for real-time monitoring of proteolytic cleavage, capable of measuring the activity of medically relevant proteolytic enzymes. For example, it could be applied for measurements of the activity of ADAMTS-13 which is an important enzyme involved in hemostasis and for which a sufficiently selective and sensitive assay is currently not available.20,21 In this paper we investigated the effect of plasmonic coupling in a model system for a proteolytic cleavage assay, containing the tryptophan–Tb3+ FRET pair and AgPr.
Experimental
Protein solutions and chemicals
Trypsin from Porcine pancreas was purchased from Sigma. Solutions of both trypsin and LBTtrp (synthesized in-house) were prepared by dissolving the protein powder in 10 mM tris HCl buffer pH 7.5. Trypsin and LBTtrp concentrations were estimated using absorption spectroscopy measuring the absorbance at 280 nm and using the molar extinction coefficient of 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−122 for trypsin and the computed molar extinction coefficient of 5625 M−1 cm−1 by ProtParam23 for LBTtrp. Terbium(III) chloride hexahydrate was purchased from Strem Chemicals. Stock solution of terbium(III) chloride hexahydrate was prepared by dissolving the powder in deionized water. Tris HCl buffer was prepared using Trizma base from Sigma and pH adjusted by adding 5 M HCl. Trisodium citrate, poly(sodium 4-styrene sulfonate), sodium borohydrate, and silver nitrate were purchased from Sigma. All chemicals were used as supplied without further purification.
700 M−1 cm−122 for trypsin and the computed molar extinction coefficient of 5625 M−1 cm−1 by ProtParam23 for LBTtrp. Terbium(III) chloride hexahydrate was purchased from Strem Chemicals. Stock solution of terbium(III) chloride hexahydrate was prepared by dissolving the powder in deionized water. Tris HCl buffer was prepared using Trizma base from Sigma and pH adjusted by adding 5 M HCl. Trisodium citrate, poly(sodium 4-styrene sulfonate), sodium borohydrate, and silver nitrate were purchased from Sigma. All chemicals were used as supplied without further purification.
Preparation of silver prisms
Silver nanoparticle seeds were synthesized by preparing a solution of 2.5 mM trisodium citrate, 14 μM poly(sodium 4-styrene sulfonate) and 60 μM sodium borohydrate. To the freshly prepared solution 5 mL 500 μM silver nitrate was added dropwise (approximately 1 mL min−1) with constant stirring at room temperature. The final silver nitrate and sodium borohydrate ratio was 1:4.
500 μL of the silver seed solution was slowly transferred into 6 mL 150 μM ascorbic acid under constant stirring. The synthesis of AgPr was carried out by dispensing 3 mL freshly prepared 500 μM silver nitrate from a buret (0.1 mL min−1) with constant stirring. 3 batches of AgPr were prepared and mixed after the synthesis. The final AgPr solution was characterized using UV-VIS spectroscopy, atomic force microscopy (AFM) and Nanosight Tracking Analysis (NTA).
LBTtrp peptide synthesis
LBTtrp (CCGAKSAFIDTNNDGWIEGDELLE) was synthesized in an automated solid-phase peptide synthesizer from Activotec (Activo P11) on rink amide MBHA resin (Advanced ChemTech) with Fmoc protected amino acids (Advanced ChemTech) producing amidated peptides. The peptide was cleaved from the resin by the addition of 3 mL cleavage solution (2.5% TIS, 2.5% EDT, 2.5% Milli-Q water and 92.5% TFA). The cleavage reaction was carried out for 60 min under continuous shaking and the resin was washed with 3 mL TFA. The peptide was precipitated by the addition of 10 mL ice-cold diethylether. The sample was centrifuged for 10 min at 4500 rcf and the supernatant decanted. The precipitate was freeze-dried, flushed with argon and stored at −20 °C.3D Peptide structure modeling with Yasara and peptide sequence analysis with ProtParam
The 3D structure of the synthesized LBTtrp peptide was predicted with the help of the modeling program Yasara. The final model of LBTtrp was energy minimized with the Amber force field in Yasara. The obtained 3D structure was displayed using RasWin molecular Graphics Windows Version 2.7.5.2. Distances between Tb3+ and the Trp residue, Tb3+ and the SH-group of Cys2, and between Trp residue and the SH-group of Cys2 were estimated using the “monitor tool” in RasWin molecular Graphics Windows Version 2.7.5.2. ProtParam provided by ExPASy was used to predict physico-chemical parameters of the LBTtrp peptide sequence.Immobilization of LBTtrp onto AgPr and uncoupling by trypsin cleavage
For each characterization technique, the following immobilization procedure was performed. A sample of 10 μM LBTtrp in 10 mM tris HCl pH 7.5 chelated with 196 μM Tb3+ in the presence of 0.01 nM AgPr was incubated for 1.5 hours at room temperature under constant stirring. The same molar ratio between LBTtrp, Tb3+ and AgPr was used for each characterization technique. The sample was characterized immediately after incubation either using steady state fluorescence spectroscopy or using transient absorption spectroscopy.Cleavage of the LBTtrp–Tb3+ peptide attached to the AgPr was conducted in the presence of 1:40 (w/w) trypsin for 2 hours at room temperature under constant stirring. The sample was characterized immediately after incubation, either using steady state fluorescence spectroscopy or using transient absorption spectroscopy.
For localized plasmon resonance (LPR) detection using UV-VIS spectroscopy the uncoupled LBTtrp–Tb3+ as well as trypsin were removed from the solution by centrifugation at 4500 rcf for 10 min. After centrifugation the supernatant was decanted and the pellet was resuspended in buffer (initial volume).
UV-VIS absorption spectroscopy
A thermo scientific UV-VIS spectrophotometer (VWK International UV1 v4.60, West Chester, PA) was used to characterize synthesized AgPr, LBTtrp peptide, and peptide coated AgPr. The path length of the quartz cuvette was 1 cm and the acquired absorbance spectra were acquired in the range of 220–850 nm.AFM
NT-MDT NTEGRA Aura multimode AFM was used to estimate the particle size and visualize the shape of the AgPr. The microscope was operated in intermittent contact mode using OMCL-AC160TS cantilevers (Olympus, k = 42 N m−1, f = 300 kHz). A drop of AgPr solution was dried overnight on a mica wafer. The microscope was controlled using NTEGRA software Nova 1.0.26 RC1 and the captured images were plane subtracted in the program.Nanoparticle Tracking Analysis (NTA)
NTA measurements were performed using a NanoSight LM12 (NanoSight, Salisbury, United Kingdom), equipped with a 640 nm laser. AgPr stock solution was diluted 10 times and injected into the sample chamber until the chamber was entirely filled without air bubbles. All measurements were performed at room temperature and the precise temperature monitored in the sample chamber. The software used for recording and analyzing the images was the NTA 2.1. The motion of AgPr in solution was recorded and tracked for 120 s using a manual shutter and gain adjustments. Three measurements of the AgPr batch were performed. The mean size and standard deviation (SD) were calculated using the NTA software based on the sizes of all the particles analyzed (total number of tracks = 17870).
Steady state fluorescence
Fluorescence characterization of 10 μM LBTtrp in the absence and the presence of 196 μM Tb3+ and 0.01 nM AgPr was carried out using a RTC 2000 PTI spectrofluorimeter at room temperature. Also, fluorescence measurements of Tb3+ chelated LBTtrp coupled to AgPr were performed before and after trypsin cleavage of the peptide construct. The slit widths were set to 5 nm and the quartz cuvette path length was 1 cm. Intrinsic LBTtrp fluorescence emission spectra and Tb3+ luminescence spectra were acquired with an excitation at 280 nm. The spectra were acquired in the range of 320–450 and 570–610 nm.Transient absorption
The transient absorption studies were performed using a femtosecond laser system and a commercial transient absorption spectrometer (Helios, Ultrafast Systems LLC). The femtosecond oscillator was a mode locked Tsunami Ti:sapphire femtosecond laser (Spectra-Physics) pumped by a Millennia V diode laser. The ∼80 fs pulses from the Tsunami were used to seed a chirped-pulse regenerative amplifier (Spectra-Physics, Spitfire, pumped by a high-power diode laser, Evolution, also from Spectra-Physics), yielding 100 fs pulses with 1 mJ of energy per pulse at a repetition rate of 1 kHz (centered at 800 nm). The output of the amplifier was divided into a high (95%) and a low (5%) power beam used to generate the pump and probe pulses, respectively. The probe beam was generated inside the Helios by white light continuum generation in a sapphire plate. The white light spectrum covered the range 420–700 nm. The 280 nm pump pulses were generated by frequency doubling the output of an optical parametric amplifier (Topas-White-SHS, Light Conversion). This resulted in a pump beam with an average power of ∼2 mW at a repetition rate of 1 kHz. The spectral width of the UV pump pulses was approximately 10 nm FWHM. The pump beam was passed through an optical delay line and a chopper at 500 Hz inside the Helios. Every second pump pulse was blocked by the chopper and the transient absorption with and without pump was recorded by the Helios. The transient absorption setup is depicted in Fig. 2.
 | ||
| Fig. 2 A schematic of the transient absorption spectroscopy setup. The ultrafast amplified laser source consists of 6 components: a 532 nm continuous wave pump laser (Millenia V), a mode-locked titanium:sapphire laser (Tsunami), a 527 nm nanosecond pump laser (Evolution) and an amplification system (Spitfire). The majority of the power from the amplifier was used to generate 280 nm pump pulses inside an optical parametric amplifier (TOPAS-White-SHS), while a small part of the power was used for generation of broadband probe pulses by white light continuum generation in a sapphire plate inside the transient absorption spectrometer (Helios). The optical delay line inside the transient absorption spectrometer allowed the probe pulse to be delayed with respect to the pump pulse. | ||
The transient absorption spectra were measured for time delays between 545 ps and 3300 ps, and each sample was scanned 3 times and averaged. The transient absorption spectra were recorded using the software program supplied with the Helios absorption spectrometer.
The LBTtrp peptide concentration used for transient absorption experiments was 114 μM in 10 mM tris HCl pH 7.5 buffer. The sample was placed in a quartz cuvette with a path length of 0.1 cm. Transient absorption spectra were acquired before and after the addition of Tb3+. Subsequently another set of transient absorption measurement was performed after AgPr addition. Transient absorption measurements were also acquired for the following solutions: (1) 114 μM LBTtrp peptide with 0.07 nM AgPr, (2) 10 mM tris HCl pH 7.5, (3) 2.1 mM Tb3+ in buffer, (4) 0.07 nM AgPr in buffer, (5) 114 μM Tb3+ with 0.07 nM AgPr. The scans were carried out at 21 ± 1 °C and the samples were continuously stirred in order to ensure homogeneous absorption of the biomolecules in solution.
Data analysis
| y = A1 × exp(−x/τ1) + y0, | (3) |
Results and discussion
AgPr synthesis and characterization
AgPr were synthesized by silver nitrate reduction as described in the Experimental section. A typical AFM image of AgPr showing the shape of the particles is depicted in Fig. 3. | ||
| Fig. 3 AFM image of AgPr deposited onto a mica substrate. The estimated average size of the AgPr is ∼50 nm in width and ∼6 nm in height. | ||
The obtained particles were flat prisms with a predominant triangular shape and a typical size of ∼50 nm in width and ∼6 nm in height as observed using AFM. NTA revealed an average size of 33 ± 1 nm. The average size difference between AFM and NTA can be explained by the fact that the NTA program estimates a real time spherical particle size distribution in solution based on the Stokes–Einstein equation. The analyzed solution contained prism shaped nanoparticles, which would lead to a size under-estimation due to the non-spherical shape of the AgPr. Nanoparticle stock solution concentration was estimated using NTA to be approximately 3.4 × 1014 particles per L.
LBTtrp 3D structure and the biosensor model
The physico-chemical properties of LBTtrp have been estimated by ProtParam on the basis of the fasta sequence: CCGAKSAFIDTNNDGWIEGDELLE revealing an isoelectric point of 3.71. The molecular weight of the peptide was calculated to be approximately 2600 g mol−1 while the extinction coefficient at 280 nm was estimated to be 5625 M−1 cm−1. A schematic representation of the biosensor model system used to study the energy transfer between Trp, Tb3+ and AgPr is displayed in Fig. 4. The LBTtrp–Tb3+ structure in this figure is based on the modeled structure in Yasara. | ||
| Fig. 4 Schematic of the biosensor model. AgPr is the silver nanoprism. The coupling between the AgPr and the LBTtrp peptide is achieved via two cysteine residues. The trypsin cleavage site is located after the lysine residue and 5 amino acids away from the N-terminus of the peptide. Tb3+ is coordinated with the lanthanide binding tag consisting of 17 amino acids. The sensitizer, Trp, is depicted in ball and stick and Tb3+ as a sphere. The arrows indicate the studied interactions. The AgPr in the model is not drawn to scale. | ||
Distances between Tb3+ and the Trp residue, Tb3+ and the SH-group of Cys2, and between Trp residue and the SH-group of Cys2 were measured using the Rasmol “monitor tool”. Distances in the model were measured to be 7 Å, 31 Å and 33 Å, respectively.
Verification of LBTtrp coupling and uncoupling onto AgPr
Synthesized AgPr were analyzed using UV-VIS absorption spectroscopy and revealed a single absorption band centered at ∼640 nm as shown in Fig. 5. The absorption peak can be assigned to the localized plasmon resonance of AgPr, which is in accordance with results found in the literature.24 | ||
| Fig. 5 Normalized absorption spectra of AgPr (triangles), LBTtrp–Tb3+ coupled to AgPr (stars) and LBTtrp–Tb3+–AgPr cleaved with trypsin (squares). | ||
Upon LBTtrp–Tb3+ coupling to the AgPr surface, via the engineered thiol/silver linker, a 45 nm red-shift was observed in the LPR absorption peak of AgPr. Trypsin cleavage of the peptide uncoupled the lanthanide binding moiety of the LBTtrp–Tb3+ peptide from AgPr resulting in a blue shift of the LPR absorption maximum of approximately 32 nm. As can be observed in Fig. 5, the LPR absorption maximum of the AgPr decoupled from the LBTtrp peptide by trypsin displays a 13 nm red-shift indicating that the amino acids before the trypsin cleavage site in LBTtrp (CCGAK) remain attached to AgPr via the two thiol groups. Moreover, LBTtrp binding to AgPr is preventing AgPr aggregation in solution upon Tb3+ addition (data not shown). These results are in accordance with the literature where several studies have confirmed the correlation between LPR absorption band shifts and changes in refractive index upon biomolecular coupling onto nanoparticle surfaces.25 In fact, several optical nanobiosensors based on LPR absorption band changes upon biomolecular coupling to the nanoparticle surface are already used in various fields such as drug design, clinical diagnostics and environmental control.2 The focus in this study is on the metal–Trp–Tb3+ interactions in the LBTtrp–Tb3+–AgPr complex and its potential applications for biosensing.
Metal–Trp–Tb3+ interactions in the LBTtrp–Tb3+–AgPr complex
Fluorescence spectroscopy measurements were performed in order to investigate the effects of AgPr on the Trp fluorescence and Tb3+ luminescence upon LBTtrp–Tb3+ immobilization to AgPr. The measured Tb3+ luminescence spectra in the range coinciding with the AgPr LPR absorption peak are shown in Fig. 6. | ||
| Fig. 6 Tb3+ luminescence upon sensitizer (Trp) excitation at 280 nm. The spectra show Tb3+ luminescence of free LBTtrp (squares), LBTtrp chelated with Tb3+ (circles), LBTtrp–Tb3+ coupled to AgPr (triangles) and LBTtrp–Tb3+ uncoupled from AgPr by trypsin cleavage of the LBTtrp peptide (stars). | ||
Trp fluorescence of LBTtrp shows a noticeable decrease of 40% upon Tb3+ binding by LBTtrp, while Tb3+ luminescence at the same time increases (Fig. 6 and 7) confirming the sensitizing effect of Trp on Tb3+, as has been stated before.26
Immobilization of LBTtrp–Tb3+ on AgPr leads to a 9% Tb3+ luminescence decrease, while decoupling of the Tb3+ binding moiety by trypsin cleavage from the AgPr surface yields a 22% increase in Tb3+ luminescence (Fig. 6 and 8A). No significant effect on Trp fluorescence was observed upon LBTtrp–Tb3+ coupling to AgPr, whereas uncoupling by trypsin leads to a 17% decrease in Trp fluorescence emission intensity (Fig. 7 and 8B). These data suggest that plasmonic coupling between Tb3+ and AgPr has a quenching effect on the Tb3+ excited state, while only a negligible quenching by AgPr was observed on the Trp excited state. In addition, upon uncoupling of LBTtrp–Tb3+ from the AgPr Trp, fluorescence decreased while Tb3+ luminescence increased (Fig. 6 and 7). This indicates that the energy transfer between Trp and Tb3+ was improved compared to free LBTtrp–Tb3+. Since the same increase in Tb3+ luminescence and the decrease in Trp fluorescence were observed when cleaving free LBTtrp–Tb3+ (data not shown), the observed change is most likely related to conformational rearrangements in the LBTtrp peptide upon cleavage.
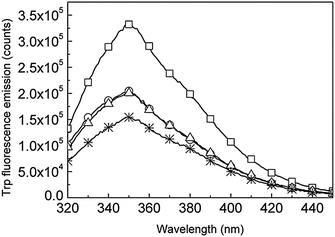 | ||
| Fig. 7 Trp fluorescence emission spectra upon 280 nm excitation of free LBTtrp (squares), Tb3+-bound LBTtrp (circles), LBTtrp–Tb3+ immobilized onto AgPr (triangles) and LBTtrp–Tb3+ uncoupled from AgPr by trypsin cleavage of the peptide (stars). | ||
 | ||
| Fig. 8 Normalized Tb3+ luminescence (A) and normalized Trp fluorescence emission (B) upon excitation at 280 nm of free LBTtrp (blank), Tb3+-bound LBTtrp (vertically hatched), LBTtrp–Tb3+ immobilized on AgPr (cross hatched) and LBTtrp–Tb3+ uncoupled from AgPr by trypsin cleavage of the peptide (grey). | ||
In LBTtrp–Tb3+, Trp is positioned approximately 7 Å away from the chelated Tb3+ ion, and thus enables intermolecular energy transfer to Tb3+, giving rise to Tb3+ luminescence. As already mentioned, several scientific papers have been published on the energy transfer between lanthanides and various types of sensitizers.27 For a range of different sensitizers the energy transfer process between the sensitizer and the lanthanide ion has been described as energy transfer via the triplet excited state of the sensitizer to the chelated lanthanide ion,26 as depicted in Fig. 1.
In order to study the behavior of the singlet Trp excited state (1Trp*) in the presence of chelated Tb3+ and AgPr, transient absorption measurements were performed. The results are summarized in Table 1.
| 1Trp* @ 560 nm (τ/ps) | R 2 | |
|---|---|---|
| LBTtrp | 99 ± 9 | 0.97 |
| LBTtrp Tb3+ | 37 ± 6 | 0.86 |
| LBTtrp Tb3+ AgPr | 28 ± 4 | 0.91 |
| LBTtrp AgPr | 111 ± 7 | 0.98 |
The transient absorption spectra of free LBTtrp displayed a distinct transient absorption peak at 560 nm with a lifetime of 99 ps. Both the lifetime and the position of the absorption maximum are in agreement with photolytic studies of 1Trp* found in the literature. The particular transient absorption peak at 560 nm, with a lifetime of approximately 400 ps, has previously been assigned to the transient absorption of aqueous 1Trp*.13,14 This study focuses on 1Trp* in protic environments and therefore the transient absorption lifetime of 1Trp* is expected to be shorter.28 As depicted in Table 1, Tb3+ binding to the LBTtrp peptide leads to a significant decrease in 1Trp* lifetime, but coupling of LBTtrp to AgPr does not, within the accuracy of the measurements, displays any further decrease in lifetime. These results are consistent with the observation that the immobilization of LBTtrp to AgPr in the absence of Tb3+ causes no significant change in 1Trp* lifetime. Regarding the energy transfer between Trp and Tb3+, the observed decrease in 1Trp* lifetime upon Tb3+ binding to LBTtrp can be interpreted in several ways: (1) increased non-radiative decay of 1Trp*, (2) increased direct energy transfer between 1Trp* and Tb3+, (3) the increased rate of intersystem crossing between 1Trp* and 3Trp, which could indirectly increase energy transfer to Tb3+ from the 3Trp. If the non-radiative decay rate of 1Trp* increases this event should suppress photo sensitisation of Tb3+. However, efficient photo sensitisation of Tb3+ was indeed observed. In principle, both the second and the third possibility agree with the achieved data. If the energy transfer occurs between 3Trp and Tb3+, the lifetime of 3Trp should decrease upon Tb3+ binding. A second transient absorption peak was observed at ∼450 nm, which has previously been assigned to 3Trp.13 A minor difference in 3Trp transient absorption was indeed observed between LBTtrp with and without Tb3+, but the signal to noise ratio at this wavelength was too low to determine whether the lifetime of 3Trp was significantly decreased. In summary, the lifetime of 1Trp* is influenced by Tb3+ chelation but no significant quenching of the 1Trp* by the AgPr was observed. These results support the interpretation of the steady state fluorescence studies that energy transfer occurs between Trp and Tb3+ and that the AgPr quenches Tb3+ luminescence but not Trp fluorescence emission.
Conclusions
The presented study demonstrates a responsive system based on AgPr and an engineered lanthanide binding peptide containing chelated Tb3+ with Trp as the sensitizer. The energy transfer between Trp and Tb3+ has been investigated using fluorescence spectroscopy and transient absorption spectroscopy. Coupling of LBTtrp–Tb3+ to AgPr was confirmed by an induced red-shift in the LPR absorption band. The detachment of the lanthanide binding moiety of the peptide from AgPr by tryptic digest was verified by a blue-shift of the LPR absorption band. When LBTtrp–Tb3+ was coupled to AgPr a 9% decrease in Tb3+ luminescence was observed due to quenching. Subsequent decoupling of the lanthanide moiety from AgPr by trypsin increased the Tb3+ luminescence by 22%.This demonstrates the potential for applying the investigated system as a sensitive optical biosensor for enzymatic activity detection, as well as a sensitive diagnostic system for changes in enzymatic activities. The presented system is a demonstration of a plasmonic based biosensor in solution.
Notes and references
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578 CrossRef CAS
.
- B. Sepúlveda, P. C. Angelomé, L. M. Lechuga and L. M. Liz-Marzán, Nano Today, 2009, 4, 244 CrossRef
- P. Baptista, E. Pereira, P. Eaton, G. Doria, A. Miranda, I. Gomes, P. Quaresma and R. Franco, Anal. Bioanal. Chem., 2008, 391, 943 CrossRef CAS
- A. J. Haes, S. Zou, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. B, 2004, 108, 109 CrossRef CAS
- J. L. Lyon, D. A. Fleming, M. B. Stone, P. Schiffer and M. E. Williams, Nano Lett., 2004, 4, 719 CrossRef CAS
- H. Imahori and S. Fukuzumi, Adv. Biomater., 2001, 13, 1197 CAS
- N. Soh, Sensors, 2008, 8, 1004 CrossRef CAS
- P. R. Selvin, IEEE J. Sel. Top. Quantum Electron., 1996, 2, 1077 CrossRef CAS
- H. Edelhoch, Biochemistry, 1967, 6, 1948 CrossRef CAS
- E. I. Onstott and C. J. Brown, Anal. Chem., 1958, 30, 172 CrossRef CAS
- S. Pandya, J. Yu and D. Parker, Dalton Trans., 2006, 2757 RSC
- B. R. Sculimbrene and B. Imperiali, J. Am. Chem. Soc., 2006, 128, 7346 CrossRef CAS
- D. Creed, Photochem. Photobiol., 2008, 39, 537–562 CrossRef
- H. Kandori, R. F. Borkman and K. Yoshihara, J. Phys. Chem., 1993, 97, 9664–9667 CrossRef CAS
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171 CrossRef CAS
- E. Matveeva, Z. Gryczynski, J. Malicka, I. Gryczynski and J. R. Lakowicz, Anal. Biochem., 2004, 334, 303 CrossRef CAS
- I. Gryczynski, E. G. Matveeva, P. Sarkar, S. Bharill, J. Borejdo, W. Mandecki, I. Akopova and Z. Gryczynski, Chem. Phys. Lett., 2008, 462, 327 CrossRef CAS
- N. Akbay, J. R. Lakowicz and K. Ray, J. Phys. Chem. C, 2012, 116, 10766 CAS
- K. Ray, R. Badugu and J. R. Lakowicz, Langmuir, 2006, 22, 8374 CrossRef CAS
- F. Peyvandi, R. Palla, L. A. Lotta, I. Mackie, M. A. Scully and S. J. Machin, J. Thromb. Haemostasis, 2010, 8, 631 CrossRef CAS
- J. D. Studt, M. Bohm, U. Budde, J. P. Girma, K. Varadi and B. Lammle, J. Thromb. Haemostasis, 2003, 1, 1882 CrossRef CAS
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411 CrossRef CAS
-
E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, The Proteomics Protocols Handbook, Humana Press Inc., 2005, pp. 571–607 Search PubMed
- A. J. Frank, N. Cathcart, K. E. Maly and V. Kitaev, J. Chem. Educ., 2010, 87, 1098 CrossRef CAS
- P. Englebienne, A. Van Hoonacker and M. Verhas, Spectroscopy, 2003, 17, 255 CrossRef CAS
- M. H. V. Werts, J. W. Verhoevena and J. W. Hofstraat, J. Chem. Soc., Perkin Trans. 2, 2000, 433 RSC
- H. E. Rajapakse, D. R. Reddy, S. Mohandessi, N. G. Butlin and L. W. Miller, Angew. Chem., Int. Ed., 2009, 48, 4990 CrossRef CAS
- Y. Chen, B. Liu, H. T. Yu and M. D. Barkley, J. Am. Chem. Soc., 1996, 118, 9271 CrossRef CAS
| This journal is © the Owner Societies 2013 |