In situ spectroscopic investigation of oxidative dehydrogenation and disproportionation of benzyl alcohol†
Received
15th February 2013
, Accepted 2nd May 2013
First published on 2nd May 2013
Abstract
In the solvent free oxidation of benzyl alcohol, using supported gold–palladium nanoalloys, toluene is often one of major by-products and it is formed by the disproportionation of benzyl alcohol. Gold–palladium catalysts on acidic supports promote both the disproportionation of benzyl alcohol and oxidative dehydrogenation to form benzaldehyde. Basic supports completely switch off disproportionation and the gold–palladium nanoparticles catalyse the oxidative dehydrogenation reaction exclusively. In an attempt to provide further details on the course of these reactions, we have utilized in situ ATR-IR, in situ DRIFT and inelastic neutron scattering spectroscopic methods, and in this article we present the results of these in situ spectroscopic studies.
Introduction
Supported bimetallic catalysts have received considerable attention because of their enhanced catalytic activities compared to their monometallic analogues, due to a synergistic effect that has been observed between the two metals.1,2 They have been reported as efficient catalysts with superior selectivity and/or stability for a variety of reactions including selective oxidation,2–5 hydrogenation/hydrogenolysis,6–8 direct synthesis of hydrogen peroxide9,10 and many more.1,11 Selective oxidation of benzyl alcohol to benzaldehyde is one of the most studied reactions because of the potential applications of benzaldehyde as an intermediate in the production of fine chemicals and fragrances.12–15 Achieving high selectivity to benzaldehyde has been a long term objective for this reaction. Designing catalysts to achieve high selectivity needs detailed knowledge about the origin and mechanism of the formation of by-products. Recently, we reported that 1% AuPd/TiO2, prepared by a sol immobilization method, is a catalyst for the solvent-free aerobic oxidation of benzyl alcohol.16 In a typical reaction, besides benzaldehyde, toluene, benzoic acid and benzyl benzoate are also formed,| | | 2PhCH2OH → PhCHO + PhCH3 + H2O | (I) |
lowering the selectivity of benzaldehyde. Among the by-products detected, toluene is formed in substantial amount and hence it was necessary to understand the mechanism of its formation. Toluene has been reported to be formed by the disproportionation of benzyl alcohol [eqn (I)].17,18| | 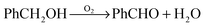 | (II) |
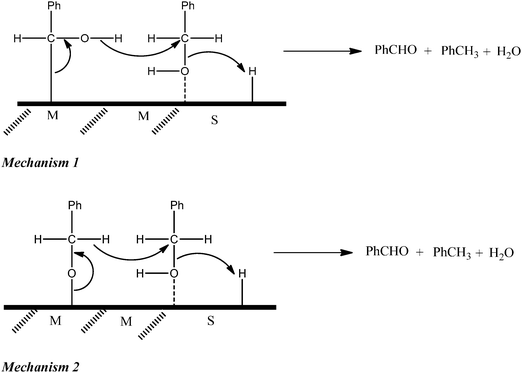
Benzaldehyde is formed by the oxidative dehydrogenation of benzyl alcohol [eqn (II)] and further oxidation of benzaldehyde results in benzoic acid. Previous kinetics studies, on the mechanism of these two reactions, have revealed that the adsorbed state for the disproportionation reaction is different from that of the oxidative dehydrogenation.18In situ spectroscopic methods are the best tools to characterize these adsorbed species over the catalyst surface.19–21 However, for the current system, this could not be done earlier because benzaldehyde is formed by both disproportionation and oxidative dehydrogenation. It is difficult to distinguish between these two sources of benzaldehyde. Recently, we reported a method to quantitatively differentiate (a) oxidative dehydrogenation and (b) disproportionation reactions when both reactions are active in the same pot.22 As a part of the study, we found that gold–palladium nanoparticles supported on basic oxides, like ZnO and MgO, switch-off the disproportionation completely.22 The extent of the oxidation reaction does not change with the change in support and hence, the active site for the oxidation reaction appears to be the metallic sites. However, the extent of the disproportionation is highly dependent on the support and the active site for the disproportionation reaction seems to be the metal–support interface.22 Possible molecular mechanisms for the disproportionation process were proposed (Scheme 1), key features of which were the modes of interaction of benzyl alcohol molecules with the catalyst/support surfaces (chemisorption on the metal through C–H or O–H bond cleavage; binding to the support involving hydrogen bonding) and the concerted nature of the bonding changes during reaction.18 For oxidative dehydrogenation, analogous mechanisms can be drawn to show the reaction of bound oxygen species with benzyl alcohol attached to the metal surface either through its benzylic C or O atom.18
These recent developments have helped us to design experiments, not possible before, where we could perform these two reactions separately, without interference from the other reaction. For example, oxidative dehydrogenation of benzyl alcohol could be performed exclusively by using ZnO or MgO supported gold–palladium catalysts under an oxygen atmosphere. Disproportionation of benzyl alcohol could be performed exclusively using TiO2 or C supported gold–palladium catalysts under anaerobic conditions. When TiO2 or C supported catalysts are used under oxygen atmosphere, benzyl alcohol undergoes oxidative dehydrogenation and disproportionation in parallel.
In situ Attenuated Total Reflection Infra-Red (ATR-IR) spectroscopy has received considerable attention as a tool for process monitoring under reaction conditions.23–25 The composition of a reaction mixture is probed by an internal reflection element (ATR sentinel). Molecules in the proximity of the sentinel's surface interact with the evanescent IR wave ranging a few μm into the probed medium resulting in molecule specific IR absorption. Implementing an IR sentinel in the bottom of an autoclave batch reactor allows for measuring in situ under conditions of up to a temperature of 350 °C and 200 bar pressure. Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectroscopy is a widely available and well-understood technique for in situ studies of heterogeneous catalysts.26–29 This technique has been widely used to characterize the chemical species adsorbed on the catalyst surface and can be used as a complimentary technique to the ATR-IR method. Inelastic Neutron Scattering (INS) Spectroscopy is a complementary form of vibrational spectroscopy, where the scattering event is between a neutron and the atomic nucleus, thus the electronic nature of the material – metal, semiconductor, or insulator – is irrelevant.30–32 The scattering intensity depends on the incoherent inelastic scattering cross section and the amplitude of vibration. For 1H both of these are large but they are small for all other elements present in this work; consequently the scattered intensity is dominated by hydrogen motion. We have carried out in situ spectroscopic studies over supported gold–palladium catalysts using these three techniques and in this paper we report our findings. Though many attempts have been made to study the mechanism of the benzyl alcohol oxidative dehydrogenation, no attempt has been made to study this disproportionation reaction. To the best of our knowledge, the unique combination of in situ ATR-IR, in situ DRIFT and INS techniques has never been attempted before.
Experimental
Materials
Benzyl alcohol (Aldrich, anhydrous >99%), TiO2 (P25 Evonik), ZnO (Sigma Aldrich), MgO (BDH), Activated C (Darco© G60, Sigma Aldrich), HAuCl4 (Sigma Aldrich) and PdCl2 (Johnson Matthey) were used without further purification. Supported gold–palladium catalysts were prepared by a sol immobilization method and the detailed procedure for preparing these catalysts is reported elsewhere.18,22 These catalysts were used without any further modifications.
In situ ATR-IR monitoring of the reactions was conducted in a 40 mL Parr stainless steel autoclave reactor equipped with a Mettler Toledo ReactIR 45m ATR-IR instrument using a diamond sentinel and a HgCdTe detector at −196 °C in the spectral range of 4000–650 cm−1.33,34 The reaction temperature was monitored and maintained using a thermocouple fitted into the reactor and connected to the controller. The reaction mixture was stirred at 1100 rpm to avoid any mass transfer limitations. The reactor vessel was charged with 15 mL of benzyl alcohol and 0.015 g of catalyst, then was closed and purged with either pure O2 or Ar thrice with constant stirring and finally pressurized to 5 bar and 20 bar, respectively. This pressure was maintained throughout the reaction time with constant replenishment in the case of O2. Heating of the reactor was started with constant stirring and ATR-IR spectra of the reaction mixture were measured every 60 s at a resolution of 4 cm−1. However, for clarity, only selected spectra are presented here. Difference spectra were created by subtraction of a pure benzyl alcohol reference spectrum acquired at 120 °C under 20 bar Ar pressure from the in situ reaction spectrum in liquid phase. Time on-line profiles to follow benzyl alcohol consumption and benzaldehyde production at 120 °C were constructed by integration of the 1016 and 1039 cm−1 double band (integration limits 1060–950 cm−1, baseline 1070–930 cm−1) and the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band at 1703 cm−1 (integration limits 1730–1670 cm−1, baseline 1740–1660 cm−1), respectively. The species present in the liquid phase (mostly) are characterized by this technique. For ATR-IR studies, we have used 1%AuPd/MgO catalyst whereas for other studies we have used 1%AuPd/ZnO catalyst. Our earlier studies have shown that, mechanistically these two catalysts behave in a similar way and thus could be swapped for the mechanistic investigation.
DRIFT spectroscopy studies were performed using a Bruker Tensor 27 spectrometer fitted with a HgCdTe (MCT) detector and a Harrick Praying Mantis HVC-DRP-4 cell equipped with ZnSe windows. Samples were mixed with benzyl alcohol and added to the cell. An alternative method of adding benzyl alcohol was the use of an in-line saturator heated to 150 °C. After an initial scan at room temperature the sample was heated to 280 °C at 1 °C min−1 under an atmosphere of flowing N2 20 ml min−1 or O2 20 ml min−1 (flows regulated by Bronkhorst mass flow meters). Spectra were run in the range of 4000–600 cm−1, with 64 accumulation scans, every 10 °C. Difference spectra were used with catalysts prior to the addition of benzyl alcohol being used as the background. This removed interference from bands associated with PVA used as a stabilizing agent in catalyst preparation. Therefore, all the spectra observed were from that of adsorbed species on the catalyst surface.
O) band at 1703 cm−1 (integration limits 1730–1670 cm−1, baseline 1740–1660 cm−1), respectively. The species present in the liquid phase (mostly) are characterized by this technique. For ATR-IR studies, we have used 1%AuPd/MgO catalyst whereas for other studies we have used 1%AuPd/ZnO catalyst. Our earlier studies have shown that, mechanistically these two catalysts behave in a similar way and thus could be swapped for the mechanistic investigation.
DRIFT spectroscopy studies were performed using a Bruker Tensor 27 spectrometer fitted with a HgCdTe (MCT) detector and a Harrick Praying Mantis HVC-DRP-4 cell equipped with ZnSe windows. Samples were mixed with benzyl alcohol and added to the cell. An alternative method of adding benzyl alcohol was the use of an in-line saturator heated to 150 °C. After an initial scan at room temperature the sample was heated to 280 °C at 1 °C min−1 under an atmosphere of flowing N2 20 ml min−1 or O2 20 ml min−1 (flows regulated by Bronkhorst mass flow meters). Spectra were run in the range of 4000–600 cm−1, with 64 accumulation scans, every 10 °C. Difference spectra were used with catalysts prior to the addition of benzyl alcohol being used as the background. This removed interference from bands associated with PVA used as a stabilizing agent in catalyst preparation. Therefore, all the spectra observed were from that of adsorbed species on the catalyst surface.
Inelastic neutron scattering (INS) spectroscopy
INS spectra were recorded with the MAPS spectrometer at ISIS; the instrument is described in detail elsewhere.32 In brief, a single energy, Ei, is selected from the incident, pulsed polychromatic beam by a Fermi chopper; after scattering from the sample, the scattered neutrons are detected by a large area (∼16 m2) of 3He detectors. For these measurements MAPS was run with the high resolution slit package (∼1.5% Ei) in the Fermi chopper. Incident energies of 4033 cm−1 (600 meV), 2017 cm−1 (250 meV) and 484 cm−1 (60 meV) were used to record the O–H/C–H stretch region, the fingerprint region and the region where framework motions (torsions, C–C–C bend) occur. The catalyst (8 g) was placed inside Conflat sealed, flow through Inconel cans between quartz wool.35 Then the inlet line of the can was connected with gas (He) and left for period of 12 h at a temperature of 120 °C to remove any absorbed species. Benzyl alcohol vapour was dosed onto the surface of the catalyst at room temperature (25 °C) for a period of 12 h. The system for dosing benzyl alcohol consists of a He line going through a heated benzyl alcohol (150 °C) reservoir and MAPS sample cell kept at 25 °C. The cans were then loaded into a closed cycle refrigerator in MAPS, cooled to <−253 °C and the spectra recorded for 6–8 h at each of the three incident energies. All species chemisorbed species or strongly adsorbed on to the solid catalyst surface are characterized by this technique.
Results and discussion
The transformations of benzyl alcohol over 1%AuPd/TiO2, 1%AuPd/MgO and 1%AuPd/C catalysts were monitored using in situ ATR-IR reactor under both aerobic and anaerobic conditions to study (a) disproportionation (b) oxidative dehydrogenation reactions. In situ ATR-IR spectroscopic investigation, using the ATR-IR probe fitted in a stirred autoclave reactor, yields mostly information about the liquid phase composition and intermediates during the course of the reaction.33,34Fig. 1 displays the absorbance (difference) spectra, with respect to pure benzyl alcohol (120 °C, 20 bar Ar), during the course of the reaction using 1%AuPd/TiO2, 1%AuPd/MgO and 1%AuPd/C catalysts under both anaerobic (120 °C, 20 bar Ar) and aerobic (120 °C, 5 bar O2) conditions. Positive band(s) in the spectra indicate an increase in the concentration of the component(s) corresponding to the band and negative band(s) indicate a decrease in concentration of the component(s) corresponding to the band. All spectra show characteristic bands assignable to benzyl alcohol (substrate), benzaldehyde and water (products). Vibrational bands corresponding to toluene are largely masked by both benzyl alcohol and benzaldehyde bands and hence it was not possible to observe toluene using this in situ ATR-IR spectroscopic method.23,24 However, GC and GC-MS studies of the reaction mixtures, reported earlier, clearly indicate the formation of toluene.18,36,37 In this article the discussion is limited to non-overlapping bands and their corresponding components such as benzyl alcohol and benzaldehyde only. For 1%AuPd/TiO2, under anaerobic conditions, disproportionation of benzyl alcohol is partially observed because of the bands around 1703 cm−1, corresponding to benzaldehyde (Fig. 1a and b). As mentioned above toluene could not be observed by any characteristic bands. Two specific bands, 1016 cm−1/1039 cm−1 bands of benzyl alcohol and 1703 cm−1 band of benzaldehyde were chosen and their areas under the band, for different time intervals, were integrated and presented in Fig. 2. It is important to note that the time in the x-axis starts when the reaction temperature reaches 120 °C. For the 1%AuPd/TiO2 catalyst, under anaerobic conditions, the area corresponding to benzaldehyde increases initially slightly and very slowly decreases with time; for the same time period there is a slight increase in the area of the band corresponding to benzyl alcohol. This observation is different from what has been seen by GC analysis for the disproportionation reaction under similar reaction conditions, where we found that there is a steady increase in the amounts of benzaldehyde and toluene at the expense of benzyl alcohol. This difference could be attributed to the formation of either hemiacetal or acetal from benzaldehyde and benzyl alcohol over gold–palladium supported on an acidic support like TiO2, however, their presence in the reaction mixture could not be verified by either GC or GC-MS.22,36 The consumption of benzaldehyde by decarbonylation to form benzene could not be ruled out; the strong absorption expected for CO could not be detected by the current instrument because of the strong absorption of the diamond sentinel in the characteristic CO stretching vibration region (1850–2150 cm−1). For the same catalyst, under aerobic conditions, (Fig. 1c and d), a substantial increase of the ν(CO) band of benzaldehyde at 1703 cm−1 and another band at 828 cm−1 was observed. Furthermore, C–H stretching vibrations of the aldehyde group located at 2818, 2736, 2697 cm−1 gain in intensity. Corresponding decrease in the area of benzyl alcohol (1016 cm−1 and 1039 cm−1) was also observed (Fig. 2). Water formation results in increase of a band at 3650 cm−1. The rate of increase in benzaldehyde band or decrease in the benzyl alcohol band, under aerobic condition, is substantially higher than that under anaerobic conditions. From previous studies it has been proven that under anaerobic conditions, the only pathway for benzyl alcohol consumption and benzaldehyde formation is disproportionation whereas under anaerobic conditions, benzaldehyde is formed by both disproportionation and oxidative dehydrogenation. However, with the present set of data, we could not distinguish between these two pathways. For AuPd/MgO supported catalyst, a remarkable feature at 3697 cm−1 showed up under both anaerobic and aerobic conditions (Fig. 1e and g). This is the ν(OHMgO) band from the catalyst support. This assignment of the hydroxyl band is confirmed by the FT-IR measurement of the fresh 1%AuPd/MgO catalyst that has a sharp band exactly at 3700 cm−1 (Fig. S1, ESI†). However, no other bands attributable to species adsorbed on the MgO support could be assigned. Under anaerobic conditions, there is no increase in the formation of the benzaldehyde after a few minutes of the start of the reaction. This initial formation has been attributed to the oxidative dehydrogenation because of the adsorbed oxygen on the catalyst surface which has been proven earlier in a study using a micro packed bed reactor. This further substantiates the fact that benzyl alcohol does not undergo any transformation over 1%AuPd/MgO catalyst in the absence of atmospheric oxygen. However, under aerobic condition, there is a steady increase in benzaldehyde concentration and a corresponding decrease in the benzyl alcohol concentration, but at a much lower rate compared to 1%AuPd/TiO2. This is because the only reaction that is active under these conditions is oxidative dehydrogenation whereas in the case of 1%AuPd/TiO2 both oxidative dehydrogenation and disproportionation reactions are active. Finally, reactions of benzyl alcohol, over 1%AuPd/C catalyst, were studied under aerobic and anaerobic conditions and results are presented in Fig. 1 (i, j for anaerobic condition, k, l for aerobic conditions). Under anaerobic conditions, in contrast to 1%AuPd/TiO2, there is a steady increase in the concentration of benzaldehyde with a corresponding decrease in the benzyl alcohol concentration. This is in line with the GC observation that benzyl alcohol undergoes disproportionation under these conditions.37 Under aerobic conditions, using the same catalyst, benzaldehyde is formed at a much higher rate (Fig. 2). From these in situ ATR-IR spectroscopic studies, the rate of formation of benzaldehyde obeys the following trend; 1%AuPd/C (aerobic) > 1%AuPd/TiO2 (aerobic) > 1%AuPd/MgO (aerobic) > 1%AuPd/C (anaerobic) > 1%AuPd/TiO2 (anaerobic) = 1%AuPd/MgO (anaerobic). Except the 1%AuPd/TiO2 (anaerobic) reaction, this trend follows the GC based activity trend.37
![In situ ATR-IR difference spectra for the reaction of benzyl alcohol using 1%AuPd/TiO2, 1%AuPd/MgO and 1%AuPd/C catalysts under anaerobic and aerobic conditions in a stirred autoclave reactor. Only 3750–2500 cm−1 [ν(O–H) and ν(C–H) regions] and 1750–750 cm−1 [fingerprint region] are presented here for clarity. Pure benzyl alcohol spectrum at 20 bar Ar, 120 °C was used as reference spectrum to calculate the difference spectra. Displayed spectra taken at 120 °C for t = 0′ (black), 30′ (dark grey), 60′ (grey), 90′ (light grey), 120′ (red). (a and b) 1%AuPd/TiO2, anaerobic; (c and d) 1%AuPd/TiO2, aerobic; (e and f) 1%AuPd/MgO, anaerobic; (g and h) AuPd/MgO, aerobic; (i and j) 1%AuPd/C, anaerobic and (k and l), aerobic. The bands (1703 cm−1 for benzaldehyde and 1016 cm−1 for benzyl alcohol) used for integration for the time on-line concentration profile (Fig. 2) are highlighted.](/image/article/2013/CP/c3cp50710f/c3cp50710f-f1.gif) |
| | Fig. 1
In situ ATR-IR difference spectra for the reaction of benzyl alcohol using 1%AuPd/TiO2, 1%AuPd/MgO and 1%AuPd/C catalysts under anaerobic and aerobic conditions in a stirred autoclave reactor. Only 3750–2500 cm−1 [ν(O–H) and ν(C–H) regions] and 1750–750 cm−1 [fingerprint region] are presented here for clarity. Pure benzyl alcohol spectrum at 20 bar Ar, 120 °C was used as reference spectrum to calculate the difference spectra. Displayed spectra taken at 120 °C for t = 0′ (black), 30′ (dark grey), 60′ (grey), 90′ (light grey), 120′ (red). (a and b) 1%AuPd/TiO2, anaerobic; (c and d) 1%AuPd/TiO2, aerobic; (e and f) 1%AuPd/MgO, anaerobic; (g and h) AuPd/MgO, aerobic; (i and j) 1%AuPd/C, anaerobic and (k and l), aerobic. The bands (1703 cm−1 for benzaldehyde and 1016 cm−1 for benzyl alcohol) used for integration for the time on-line concentration profile (Fig. 2) are highlighted. | |
 |
| | Fig. 2 ATR-IR time on line peak area profiles of benzyl alcohol (left) and benzaldehyde (right) obtained by integrating the peak area of bands at 1016 cm−1 (benzyl alcohol) and 1703 cm−1 (ν(CO) of benzaldehyde) from Fig. 1. Key: black squares: 1%AuPd/TiO2, anaerobic; red circles: 1%AuPd/TiO2, aerobic; green triangles: 1%AuPd/MgO, anaerobic; blue triangles: 1%AuPd/MgO, aerobic; magenta diamond: 1%AuPd/C anaerobic and cyan triangles: 1%AuPd/C aerobic. | |
Table 1 Vibrational modes assignment in the 1800–1300 cm−1 region at 22 °C
| Benzyl alcohol (neat) |
Benzaldehyde (neat) |
Toluene (neat) |
Benzoic acid (neat) |
1 wt% AuPd/TiO2 N2 flow |
1 wt% AuPd/ZnO N2 flow |
Assignment |
|
|
1700 |
|
1685 |
1702 |
1702 |
ν(CO) |
|
|
|
|
|
1654 |
|
ν(CO) |
| 1607 |
1599 |
1604 |
1603 |
1598 |
1598 |
ν(CC) + δ(C–H) |
| 1586 |
1584 |
|
1584 |
1584 |
1584 |
ν(CC) + δ(C–H) |
| 1496 |
|
1495 |
1497 |
1496 |
1496 |
δ(C–H) + ν(CC) |
| 1454 |
1454 |
1458 |
1454 |
1454 |
1454 |
δ(C–H) + ν(CC) |
|
|
1424 |
|
1424 |
|
1418 |
|
|
|
|
1379 |
|
1391 |
|
|
| 1369 |
|
|
|
1369 |
1370 |
δ(O–H) |
|
|
|
|
|
|
1340 |
δ(C–H) + ν(CC) |
|
|
1326 |
|
1325 |
|
|
|
|
|
|
|
|
|
1311 |
|
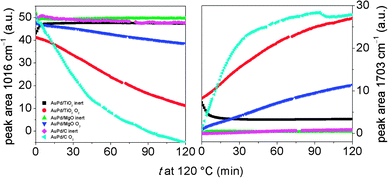
Heating of the benzyl alcohol dosed catalysts under a flow of inert gas resulted in desorption of the benzaldehyde and benzyl alcohol by 180 °C. It is important to note the presence of bands associated with irreversibly adsorbed species for both 1%AuPd/TiO2 and 1%AuPd/ZnO catalyst after heating samples to 280 °C (Fig. 3). For both catalysts, bands associated with aromatic CC and C–H modes (1592–1595 cm−1 and 1493–1496 cm−1) were observed. The 1%AuPd/ZnO catalyst had strong bands at (1533, 1419 and 1374 cm−1), indicative of benzoate species previously observed in in situ ATR infra-red studies of benzyl alcohol using Pt/Al2O3 catalysts.23 Bands associated with benzoate were also observed on 1%AuPd/TiO2, though these were considerably less intense than that observed on the ZnO catalyst. However, in addition to benzoate, a band at 1654 cm−1 with a shoulder at 1670 cm−1 was present on the 1%AuPd/TiO2 catalyst. The band at 1670 cm−1 is indicative of carbonyl bands associated with a conjugated carboxylic acid group and matches that observed for benzoic acid.23 The lower frequency 1654 cm−1 is at the low frequency extreme of the range observed for carbonyl bands and demonstrates significant electron donation to the group, weakening the CO bond. This low wavenumber carbonyl band could indicate electron donation from Au or Pd metal, showing chemisorption of the species to the metal via the carbon of the carbonyl functional group but might also arise from weak adsorption of benzaldehyde to the metal through the carbonyl O-atom [cf. 1667 cm−1 for CH3CHO on Pt(111)38]. However, it is noted that weak adsorption through the carbonyl O-atom is less likely for species still bound to the surface at 280 °C. This ambiguity in interpretation and the fact that product species are being observed means that their structure cannot be used to infer how dissociative chemisorption of benzyl alcohol occurs on the different catalysts. The proposed interaction of the metal with the carbon of the carbonyl functional group seen in the 1%AuPd/TiO2 catalyst might suggest that benzyl alcohol was chemisorbed by cleavage of a benzylic C–H bond and not the O–H bond. The reverse is true if the carbonyl oxygen of the product is attached to the metal surface and this is indicative of its precursor surface species. Both modes of interaction have been previously suggested as possible surface species.19 As the 1654 cm−1 signal was observed only for the 1%AuPd/TiO2 catalyst, which facilitates the disproportionation reaction, it suggests the importance of this chemisorbed species in the disproportionation reaction. This is re-enforced by the total absence of the 1655 cm−1 band in the basic ZnO supported catalyst which does not bring about disproportionation.
 |
| | Fig. 3 DRIFT spectra of benzyl alcohol dosed catalysts after heating the samples to 280 °C (1 °C min−1). Key: (a) 1%AuPd/TiO2 and (b) 1%AuPd/ZnO. Bands labelled are referred to in the text. | |
Inelastic neutron scattering spectroscopy
Fig. 4 and 5 show the adsorption and reaction of benzyl alcohol on 1%AuPd/TiO2 and 1%AuPd/ZnO catalyst in the C–H stretch region and Fig. 6 and 7 the corresponding spectra in the fingerprint region. Reference spectra of pure benzyl alcohol, benzaldehyde and toluene in the same regions are shown in the ESI† (Fig. S3 and S4). It can be seen that for both supports under the conditions used here, there is no adsorption of benzyl alcohol on the bare support, Fig. 4a, 5a, 6a, and 7a; the observed features are all assigned to species on the AuPd nanoparticles. The generally weak intensities observed are consistent with the low metal loading employed.
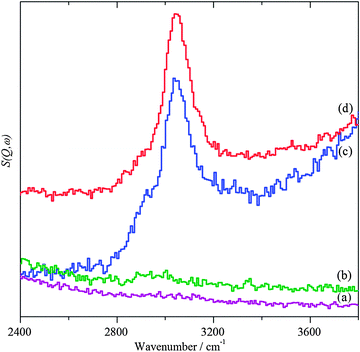 |
| | Fig. 4 INS spectra in the C–H stretch region. (a) Bare TiO2 support after dosing with benzyl alcohol, (b) AuPd/TiO2 catalyst after 20 h helium purge at 150 °C, (c) after dosing with benzyl alcohol and (d) after heating in flowing helium at 150 °C. | |
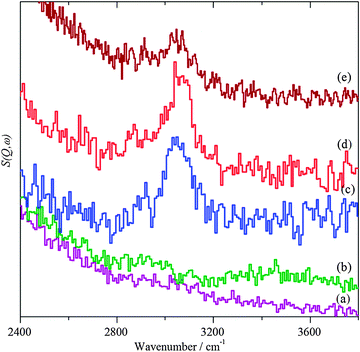 |
| | Fig. 5 INS spectra in the C–H stretch region. (a) Bare ZnO support after dosing with benzyl alcohol, (b) AuPd/ZnO catalyst after helium purge at 150 °C, (c) after dosing with benzyl alcohol, (d) after heating in flowing helium at 150 °C and (e) after reaction with O2. | |
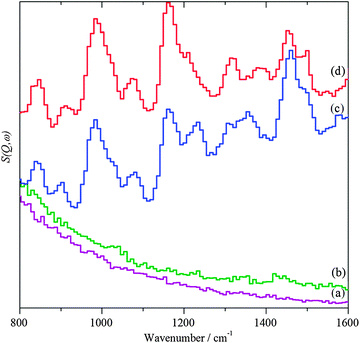 |
| | Fig. 6 INS spectra in the fingerprint region. (a) Bare TiO2 support after dosing with benzyl alcohol, (b) AuPd/TiO2 catalyst after 20 h helium purge at 150 °C, (c) after dosing with benzyl alcohol and (d) after heating in flowing helium at 150 °C. | |
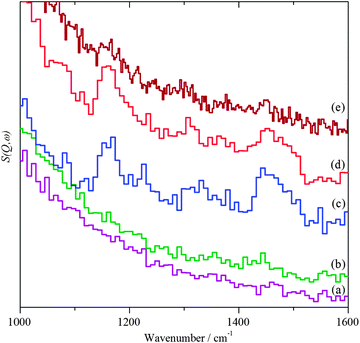 |
| | Fig. 7 INS spectra in the fingerprint region. (a) Bare ZnO support after dosing with benzyl alcohol, (b) 1%AuPd/ZnO catalyst after helium purge at 150 °C, (c) after dosing with benzyl alcohol, (d) after heating in flowing helium at 150 °C and (e) after reaction with O2. | |
From infrared and Raman spectra of solid benzyl alcohol,39 the out-of-plane C–O–H bending modes are assigned at 695 cm−1, the CH2 scissors mode at 1435 cm−1, the methylene C–H stretch modes at 2850–2950 cm−1, the aromatic C–H stretch modes at 3006–3090 cm−1 and the O–H stretch at 3170 cm−1. Ab initio calculations of the complete unit cell40 using the CASTEP code confirm these assignments and also show that the CH2 twist is at 1233 cm−1 and the in-plane C–O–H bending modes are at 1445 and 1507 cm−1, thus overlapping the CH2 scissors modes.
Exposure of the supported catalysts to benzyl alcohol results in new features in both the C–H stretch and fingerprint regions, Fig. 4c, 5c, 6c, and 7c. Comparison with the reference spectra, Fig. S3 and S4 (ESI†), suggests that this is benzyl alcohol. Overlaying the spectra of pure benzyl alcohol and that adsorbed on 1%AuPd/TiO2, Fig. 8, shows that the profile is unchanged apart from an additional weak, broad feature at ∼3210 cm−1, suggesting interaction is between the metal and the alcoholic oxygen. Curve fitting the C–H stretch region of pure benzyl alcohol, Fig. S3a (ESI†), shows that the ratio of the areas of the CH2, aromatic C–H and O–H stretch modes is 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.6:1, close to the expected30,32 ratio of 2:5:1. For benzyl alcohol adsorbed on 1%AuPd/TiO2, after allowance for the 3210 cm−1 band, the ratio is: 1.6:6:1, which given the modest signal-to-noise ratio, indicates that the molecule adsorbs intact, suggesting that the C–M and O–M species in Scheme 1 are transition states rather than intermediates. There is a small shift in the O–H stretch transition energy from 3184 down to 3172 cm−1 which would be consistent with benzyl alcohol hydrogen bonding to the neighbouring benzyl alcohol molecule. Possible assignments for the 3210 cm−1 band are to water or hydroxyls that are formed while dosing the sample at 150 °C (a temperature that is required to prevent physisorption of the benzyl alcohol on the sample). In the case of benzyl alcohol on AuPd/ZnO, the quantity of benzyl alcohol we were able to adsorb was much smaller and the signal-to-noise ratio is much poorer; however, the profile matches that of pure benzyl alcohol, again indicating that the adsorption is molecular. In this case, the 3210 cm−1 band was absent.
:6.6:1, close to the expected30,32 ratio of 2:5:1. For benzyl alcohol adsorbed on 1%AuPd/TiO2, after allowance for the 3210 cm−1 band, the ratio is: 1.6:6:1, which given the modest signal-to-noise ratio, indicates that the molecule adsorbs intact, suggesting that the C–M and O–M species in Scheme 1 are transition states rather than intermediates. There is a small shift in the O–H stretch transition energy from 3184 down to 3172 cm−1 which would be consistent with benzyl alcohol hydrogen bonding to the neighbouring benzyl alcohol molecule. Possible assignments for the 3210 cm−1 band are to water or hydroxyls that are formed while dosing the sample at 150 °C (a temperature that is required to prevent physisorption of the benzyl alcohol on the sample). In the case of benzyl alcohol on AuPd/ZnO, the quantity of benzyl alcohol we were able to adsorb was much smaller and the signal-to-noise ratio is much poorer; however, the profile matches that of pure benzyl alcohol, again indicating that the adsorption is molecular. In this case, the 3210 cm−1 band was absent.
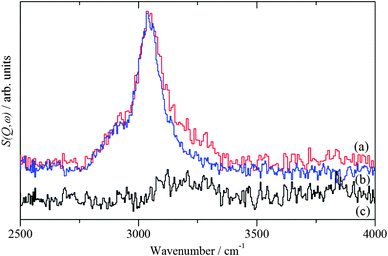 |
| | Fig. 8 Comparison of the INS spectra of (a) benzyl alcohol adsorbed on 1%AuPd/TiO2 (red trace) and (b) the pure compound (blue trace). The difference spectrum (black trace) is shown in (c) and is plotted on the same scale as (b). | |
On heating under anaerobic conditions the difference in behaviour of the two catalysts is readily apparent, in both the C–H stretch and fingerprint regions (Fig. 4d, 5d, 6d, and 7d). For the TiO2 supported catalyst, the spectral profiles in both regions change significantly. In particular, the modes at ∼1230 and 1460 cm−1, which are associated with the methylene and hydroxyl functionalities, weaken dramatically as does the signal at 2900 cm−1 assigned to the symmetric CH2 stretch; a new signal appears at 1620 cm−1. Comparison with the reference spectra shows that the changes are consistent with the loss of benzyl alcohol and formation of a mixture of toluene and benzaldehyde, although the loss of methylene signals might also be argued as suggestive of chemisorption of benzyl alcohol by benzylic C–H cleavage as well as benzaldehyde via its carbonyl oxygen. For the ZnO supported material, the profiles do not change greatly, indicating that the only effect is to remove some of the adsorbate. Heating the 1%AuPd/ZnO in oxygen resulted in reaction of the benzyl alcohol and desorption of most of the products. The very weak remnant is clearly aromatic and probably a mixture of toluene and benzaldehyde. In contrast to 1%AuPd/ZnO, where all of the reactions could be carried out on the same sample, it was necessary to prepare a separate sample for 1%AuPd/TiO2 since reaction occurs under anaerobic conditions. Benzyl alcohol was adsorbed on the catalyst; however, treatment with oxygen did not result in any change in the spectrum.
Concluding comments
It was found that benzyl alcohol undergoes competitive oxidative dehydrogenation, giving benzaldehyde, and disproportionation, to yield equimolar benzaldehyde and toluene, when treated with oxygen over an AuPd nanoparticulate catalyst supported on titania or carbon, but only oxidative dehydrogenation when the catalyst is supported on MgO or ZnO. In situ spectroscopic examination of reaction mixtures have been undertaken in an attempt to identify intermediates and surface-bound species that would throw light on the mechanisms involved. ATR-IR examination yielded signals attributable only to species in the liquid phase and, under an oxygen atmosphere, these confirmed the support-dependent differences in behaviour of the catalysts, observed previously by conventional catalytic studies. In reactions catalysed by 1%AuPd/TiO2 under argon, however, the ATR signals attributed to benzaldehyde decreased with time at the reaction temperature (120 °C) with a corresponding increase in the signal used as a measure of the benzyl alcohol remaining in the system. This deviation from expectation did not occur when 1%AuPd/C or 1%AuPd/MgO were the catalysts. A number of possible explanations of this paradoxical behaviour have been considered, the favoured one being the analytical complication introduced by the equilibrium formation of the benzyl hemiacetal or more probably acetal of benzaldehyde on the relationship between signal intensity and concentration of benzaldehyde and benzyl alcohol. A DRIFTS investigation of the same reaction systems revealed only one signal that distinguished the behaviour of the acidic (TiO2) and basic (ZnO) supports. Using 1%AuPd/TiO2 a carbonyl signal was observed at the unusually low frequency of 1655 cm−1. It has not proved possible to determine whether this arises from attachment of the carbonyl moiety to the metal through its carbonyl carbon or its carbonyl oxygen atom and thus it has not proved possible to discriminate between previously proposed modes of chemisorption of benzyl alcohol on the metal surface and hence on mechanisms for oxidative dehydrogenation. Inelastic neutron scattering has thrown somewhat greater light on the processes occurring at the catalyst surface. Benzyl alcohol appears to adsorb intact on the metal surface; a weak additional signal at 3210 cm−1 remains unassigned. It is clear, that the initial interaction of benzyl alcohol with the metal catalyst is not by benzylic C–H cleavage, although this process could be involved subsequently, giving a transient surface species that is undetectable by the present technique. We have not found any direct spectroscopic evidence for the species shown in Scheme 1 for disproportionation; those mechanistic depictions remain merely rationalisations of the organic transformations that are helpful in understanding the results from earlier isotopic labelling experiments. It is clear that even using a range of in situ vibrational spectroscopies it has proved to be difficult to unravel some of the subtle details in this fascinating reaction and those techniques using shorter timescales for observation of reaction species need to be considered.
Acknowledgements
The authors thank the EPSRC for funding, the STFC Rutherford Appleton Laboratory for access to neutron beam facilities and Dr James Pritchard and Mr Scott Davies for their assistance during INS measurements. Computing resources (time on the SCARF computer used to perform the CASTEP calculations) was provided by STFC's e-Science facility. MS thanks the European Research Council (ERC) for awarding a Marie Curie Fellowship (IEF) for Career Development (Award No. 275755), while JPH would like to express gratitude to the German Research Foundation (DFG) (postdoctoral research grant Ho4579/1-1) and the Dutch National Research School Combination Catalysis (NRSC-Catalysis) for funding.
References
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS.
- L. Prati, F. Porta, D. Wang and A. Villa, Catal. Sci. Technol., 2011, 1, 1624–1629 CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176 CrossRef CAS.
- B. Bachiller-Baeza, A. Guerrero-Ruiz, P. Wang and I. Rodriguez-Ramos, J. Catal., 2001, 204, 450–459 CrossRef CAS.
- P. D. Zgolicz, V. I. Rodriguez, I. M. J. Vilella, S. R. de Miguel and O. A. Scelza, Appl. Catal., A, 2011, 392, 208–217 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS.
- V. Dal Santo, A. Gallo, A. Naldoni, M. Guidotti and R. Psaro, Catal. Today, 2012, 197, 190–205 CrossRef CAS.
- B. E. Solsona, J. K. Edwards, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Chem. Mater., 2006, 18, 2689–2695 CrossRef CAS.
- J. M. Parera and J. N. Beltramini, J. Catal., 1988, 112, 357–365 CrossRef CAS.
-
C. H. Bamford, C. F. H. Tipper and R. G. Compton, Liquid Phase Oxidation, Elsevier, Oxford, 1980 Search PubMed.
-
R. A. Sheldon and J. K. Kochi, Metal-Catalysed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
-
M. Hudlicky, Oxidation in Organic Chemistry, American Chemical Society, Washington, DC, 1990 Search PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Bethell and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 5142–5153 RSC.
- G. Kovtun, T. Kameneva, S. Hladyi, M. Starchevsky, Y. Pazdersky, I. Stolarov, M. Vargaftik and I. Moiseev, Adv. Synth. Catal., 2002, 344, 957–964 CrossRef CAS.
- S. Meenakshisundaram, E. Nowicka, P. J. Miedziak, G. L. Brett, R. L. Jenkins, N. Dimitratos, S. H. Taylor, D. W. Knight, D. Bethell and G. J. Hutchings, Faraday Discuss., 2010, 145, 341–356 RSC.
- J. D. Grunwaldt and A. Baiker, Phys. Chem. Chem. Phys., 2005, 7, 3526–3539 RSC.
-
In Situ Spectroscopy of Catalyst, ed. B. M. Weckhuysen, American Scientific Publishers, California, 2004 Search PubMed.
- B. M. Weckhuysen, Chem. Commun., 2002, 97–110 RSC.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem.–Eur. J., 2011, 17, 6524–6532 CrossRef CAS.
- C. Keresszegi, D. Ferri, T. Mallat and A. Baiker, J. Phys. Chem. B, 2005, 109, 958–967 CrossRef CAS.
- D. Ferri and A. Baiker, Top. Catal., 2009, 52, 1323–1333 CrossRef CAS.
- J. M. Andanson and A. Baiker, Chem. Soc. Rev., 2010, 39, 4571–4584 RSC.
- S. Chilukoti, E. Widjaja, F. Gao, H. Zhang, B. G. Anderson, J. W. H. Niemantsverdriet and M. Garland, Phys. Chem. Chem. Phys., 2008, 10, 3535–3547 RSC.
- J. G. Highfield, M. Prairie and A. Renken, Catal. Today, 1991, 9, 39–46 CrossRef CAS.
- J. G. Highfield, M. H. Chen, P. T. Nguyen and Z. Chen, Energy Environ. Sci., 2009, 2, 991–1002 CAS.
- D. Ferri, M. S. Kumar, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC.
-
P. C. H. Mitchell, S. F. Parker, A. J. Ramirez-Cuesta and J. Tomkinson, Vibrational spectroscopy with neutrons, with applications in chemistry, biology, materials science and catalysis, Singapore, 2005 Search PubMed.
-
S. F. Parker, Handbook of Vibrational Spectroscopy, John Wiley & Sons, New York, 2006 Search PubMed.
- S. F. Parker, D. Lennon and P. W. Albers, Appl. Spectrosc., 2011, 65, 1325–1341 CrossRef CAS.
- J. Zakzeski, R. J. H. Grisel, A. T. Smit and B. M. Weckhuysen, ChemSusChem, 2012, 5, 430–437 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx and B. M. Weckhuysen, Green Chem., 2011, 13, 671–680 RSC.
- I. P. Silverwood, N. G. Hamilton, A. McFarlane, R. M. Ormerod, T. Guidi, J. Bones, M. P. Dudman, C. M. Goodway, M. Kibble, S. F. Parker and D. Lennon, Rev. Sci. Instrum., 2011, 82, 1–7 CrossRef.
- E. Cao, M. Sankar, E. Nowicka, Q. He, M. Morad, P. J. Miedziak, S. H. Taylor, D. W. Knight, D. Bethell, C. J. Kiely, A. Gavriilidis and G. J. Hutchings, Catal. Today, 2012, 203, 146–152 CrossRef.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem.–Eur. J., 2011, 17, 6524–6532 CrossRef CAS.
- H. Zhao, J. Kim and B. E. Koel, Surf. Sci., 2003, 538, 147–159 CrossRef CAS.
- D. A. Prystupa, A. Anderson and B. H. Torrie, J. Raman Spectrosc., 1994, 25, 175–182 CrossRef CAS.
- S. K. Nayak, R. Sathishkumar and T. N. G. Row, CrystEngComm, 2010, 12, 3112–3118 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp50710f |
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a