 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fullerene C70 characterization by 13C NMR and the importance of the solvent and dynamics in spectral simulations†
Jakub
Kaminský
*a,
Miloš
Buděšínský
a,
Stefan
Taubert
b,
Petr
Bouř
a and
Michal
Straka
*a
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo n. 2., CZE-16610, Praha 6, Czech Republic. E-mail: kaminskj@gmail.com; straka@uochb.cas.cz; Fax: +420-220183-579
bDepartment of Chemistry, Aalto University, P. O. Box 16100, FIN-00076 Aalto, Finland
First published on 19th April 2013
Abstract
The nuclear magnetic resonance (NMR) spectroscopy combined with theoretical calculations is an important tool for fullerene identification. However, the accuracy of available theoretical methods is often not adequate. Therefore, in this work, different computational aspects needed to simulate realistically chemical shifts in the C70 molecule are investigated by density functional theory (DFT) calculations. The importance of the functional choice, basis set, solvent, and molecular motions was assessed. The solvent was simulated using the implicit conductor-like polarized continuum model. The molecular motions were included via anharmonic corrections and averaging of snapshots obtained from classical and first-principles molecular dynamics (MD) simulations. Comparison to experiment revealed that density functional calculations typically overestimate the 13C NMR chemical shifts. Hybrid functionals, such as BHandH and BHandHLYP, and long-range corrected functionals, such as wB97xd and CAM-B3LYP, give the best results. While the solvent has a minor effect (chemical shift changes by ∼1 ppm), the vibrational and dynamical effects are surprisingly large, causing changes up to 9 ppm. Consideration of the latter was also necessary to explain the observed temperature dependence. While the dynamical corrections for MD performed in vacuum were overestimated, inclusion of the solvent in simulations provided more realistic results. The study thus points out the importance of an appropriate solvent model and a complex approach to the modelling, balancing the static, dynamic and environmental factors.
Introduction
Since the initial discovery of C60 by Kroto et al.1 the fullerene science has evolved into a rich multidisciplinary field. Fullerenes and their derivatives have been intensively studied as materials for solar cells, quantum computing, memory devices, magnetic storage devices, molecular containers, MRI contrast agents, and other applications.2,3 The fullerene cage structures have been rationalized by the isolated-pentagon rule (IPR).4 The number of the possible isomers of Cn grows with n. For example, the C60 fullerene has one isomer, C80 has seven isomers, and for C84, 24 IPR isomers have been predicted to exist. In spite of the similarity of different fullerenes and their isomers, owing to their high molecular symmetry, they can be in many cases distinguished according to the number and intensity of spectral lines in 13C nuclear magnetic resonance (NMR) spectra. The NMR spectroscopy, hand in hand with theoretical interpretation of the experimental NMR spectra, is thus an important analytical tool in fullerene chemistry.Identification of fullerenes can be significantly enhanced by calculations of fullerene structures and 13C NMR parameters, typically based on the density functional theory (DFT).5–11 In a review by Heine,12 however, typical accuracy of 13C NMR DFT calculations was estimated to be 2 ppm, which may not be sufficient for larger fullerenes with a denser spacing (<1 ppm) of spectral lines.
To assess the reliability of commonly used methods for calculations of the 13C chemical shift we use the C70 fullerene as a convenient model. It contains five different symmetry-equivalent carbon types (assigned as Ca–Ce in Fig. 1) which cover the range of ∼130–150 ppm spanning to a large part typical fullerene 13C chemical shifts.13 The performance of various density functionals and basis set convergence are analyzed.
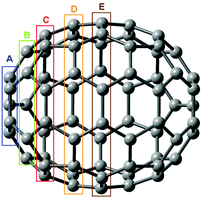 | ||
| Fig. 1 Symmetry equivalent carbon types in C70. | ||
In the next stage, we assess the effects of dynamics, temperature, and solvent on the calculated 13C NMR chemical shifts. The effects of solvent are approximated using the conductor-like polarized continuum model (CPCM).14 Several approaches of involving the molecular motion in the computations are explored: averaging snapshots obtained from classical molecular dynamics (MD) and the first-principles molecular dynamics (FPMD) trajectories, and a quantum averaging of anharmonic vibrational corrections.15,16
Methods
DFT calculations
The static DFT calculations and the first-principles MD were performed using the Turbomole 6.3.1 (ref. 17) and Gaussian 09, Revision A.02 (ref. 18) program packages. Classical MD was run using the Tinker program suite.19,20 The geometry was optimized using several DFT functionals (BP86,21,22 B3LYP,23 and wB97xd24) and the Ahlrichs-type basis sets (def2-SVP, def2-TZVP, and def2-QZVP).25,26The nuclear magnetic shielding was calculated with GGA (HCTH,27 BLYP,21,28 OLYP,28,29 BP86,21,22 PBEPBE,30 OPBE,29,30 SV5LYP28,31,32), meta GGA (M06L33), hybrid (B1LYP,34 B3LYP,23 O3LYP,35 X3LYP,36 BHandH/Gaussian09,18 BHandHLYP,18 and B9837), meta GGA hybrid (M06HF38), and range-separated hybrid (CAM-B3LYP,39 wB97xd24) functionals. The list thus enables us to compare the GGA and hybrid functionals, different exchange potentials (e.g. in B1LYP, B3LYP, O3LYP, X3LYP) and various degrees of the involvement of the HF exchange (e.g. BLYP with 0% exchange, B3LYP (20%), and BHandHLYP (50%)). Because common functionals may suffer from unphysical long-range behavior of the non-Coulomb part of exchange, the effect of long-range corrections was also taken into account by involving the CAM-B3LYP39 and wB97xd24 functionals.
The basis set convergence was investigated using the IGLO-II,40 IGLO-III,40 and IGLO-IV41,42 basis sets optimized for calculations of NMR parameters. A set of Jensen's polarization-consistent basis sets, pcS-n, was also tested.43
Calculated isotropic nuclear shielding (σ) was converted to chemical shift (δ) using tetramethylsilane (TMS) as a reference and C60 as a secondary reference, according to formula
| δi = σ(C60) − σi(C70) + δ(C60,TMS) |
Solvent modelling
Effects of solvent were approximated using the conductor-like polarized continuum model (CPCM)14 as implemented in Gaussian 09,18 using standard parameters for 1,1,2-trichloroethane. Note that 1,1,2,2-tetrachloroethane was used instead in ref. 44, not supported by Gaussian 09, but with a very similar dielectric constant (7.19 for 1,1,2-trichloroethane and 8.42 for 1,1,2,2-tetrachloroethane).Quantum vibrational averaging
The nuclear potential of C70 was expanded to a Taylor series up to fourth powers of all normal mode coordinates Qi.45 Only cubic and semi-diagonal normal mode quartic constants (with two or more identical indices) were considered, as these can be obtained by a single normal mode numerical differentiation of harmonic force fields.46 The anharmonic force field was calculated at the Hartree–Fock (HF) level using the 6-31G basis set, while the shielding derivatives were obtained at the BP86/IGLO-II level. Gaussian 0918 was used for the Hessian computations for geometries displaced in normal modes. Program S447 interfaced to Gaussian was used for the anharmonic vibrational averaging.One of the limitations of anharmonic vibrational corrections performed here is the necessity to use Hartree–Fock approximation which overestimates harmonic frequencies by about 10%.48,49 While DFT provides typically better harmonic frequencies deviating from experiment by only a few percents,50–53 it was not affordable for such extensive calculations on C70. We do not suppose that the relatively small frequency error of the HF method would significantly change the anharmonic corrections added in a perturbation way only.
For vibrational ground state ψn isotropic nuclear magnetic shielding was calculated as
| σn = 〈ψn|σ|ψn〉 |
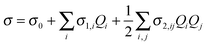
The σ1 and σ2 are the first and second normal mode isotropic shielding derivatives obtained numerically with a differentiation step of 0.2 Å. This parameter was found to be optimal for many systems including fullerenes before.54 Within the second-order degeneracy-corrected perturbational (PT2) scheme46 the wave function is expanded in the harmonic oscillator basis.16
Alternatively, the vibrational configuration interaction (VCI) was used for vibrationally averaged shieldings, where the Hamiltonian was diagonalized in the harmonic oscillator basis functions (|l〉, up to doubly excited states) limited by an interaction parameter
| |〈l|V|n〉/(En − El)| ≤ 0.1 |
A third approximation tried was based on the harmonic oscillator limit for energies55 and provided temperature-corrected shielding (TCS) as
| σ = σ0 + 0.25σiiexp(−ωi/kT)[1 − exp(−ωi/kT)]−1 |
Molecular dynamics
The BP86/def-TZVP optimized structure of C70 was minimized using the Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) procedure in Tinker. The minimized geometry was evolved for 1 ns. The classical MD trajectories with the MM3 force field56 were obtained using the modified Beeman algorithm with a 1 fs integration time step at constant temperature T = 300 K. The MM3 force field was modified for the C70 carbon atoms as described in the ESI.† Chemical shifts were calculated for 40 snapshots at the BHandHLYP/IGLO-III level and averaged. To minimize systematic error, the dynamical correction was calculated as a difference between the average and the shielding calculated for MM3-minimized geometry.First-principles molecular dynamics
FPMD simulations were performed on the BP86/def-SVP Born–Oppenheimer potential energy surface. The leapfrog Verlet algorithm as implemented in the Turbomole code17 was used to update the coordinates. To avoid problems due to the limited numerical precision in the density optimization and the force calculation, the thresholds for energy and CPHF equations convergence were tightened to 10−8 a.u. A denser numerical grid denoted as “m5” was used. The time step was 80 a.u. (1.93 fs). To achieve a better sampling of the configuration space we ran several microcanonical (NVE ensemble) dynamics trajectories 1.2 ps long. The initial velocities were randomly distributed and corresponded to the temperature of 300 K. A total of four MD trajectories of 1.2 ps were produced for the averaging of snapshots. The initial 0.2 ps part of each trajectory was discarded as an equilibration phase, and then 16 equidistant snapshots of each trajectory (64 in total) were taken for the 13C NMR calculations (BHandHLYP/IGLO-III) and averaging. To verify the time-correlation of the data the block-averaging method was used.57 The mean errors of average were found in the interval of ±0.2 to ±0.25 ppm for the NMR shielding of different carbons, which after subtraction from the C60 reference give a statistical error of ±0.35 ppm in the dynamical corrections for shielding. The dynamical corrections were obtained as differences between the average and result for the BP86/def-SVP optimized structure.NMR experiment
Fullerenes (C60 and C70) were purchased from Sigma. 13C NMR spectra of C70 were recorded in deuterated ortho-dichlorobenzene (ODCB) using a Bruker Advance II™ 600 and 500 MHz spectrometer equipped with a cryoprobe. The spectra were measured at several temperatures (278, 298, 318, 338, 358 and 378 K). A small amount of C60 was detected (at 142.86 ppm) in the sample of C70. The chemical shifts were referenced to tetramethylsilane (TMS).Results and discussion
Basis set convergence
We used optimized BP86/def-TZVP C70 structure58 to investigate the effect of the basis set and DFT functional on the 13C NMR chemical shifts in C70 calculated for the molecule at rest in vacuo. The BP86/def-TZVP level previously provided results in an excellent agreement with the experimental structural parameters for the C60 molecule.58,59Table 1 lists the 13C chemical shifts for C70 calculated using the different IGLO-n type basis sets. The IGLO-III results appear converged within 0.2 ppm as compared to the IGLO-IV basis set, which can be considered to be near the basis set limit. Corrections beyond IGLO-IV can be expected to lie below 0.2 ppm.60 Hence the error of the IGLO-III basis set calculations with respect to the basis set limit can be estimated with safe margins to be within 0.4 ppm. With a conservative error estimate of 0.4 ppm for 13C shifts the IGLO-III basis set provides a good compromise between the accuracy and the computational cost. IGLO-IV is recommended if higher accuracy is needed.
| Carbon | IGLO-II | IGLO-III | IGLO-IV |
|---|---|---|---|
| a Chemical shifts were calculated for BP86/def-TZVP optimized geometry. The BP86 functional was used for 13C chemical shifts. | |||
| Ca | 153.3 | 154.4 | 154.2 |
| Cb | 148.7 | 149.6 | 149.5 |
| Cc | 150.0 | 151.2 | 151.0 |
| Cd | 147.4 | 148.1 | 148.0 |
| Ce | 130.7 | 130.5 | 130.5 |
Following the suggestion of a referee, the basis set convergence was also tested on the set of Jensen's polarization consistent pcS-n basis sets43 known to provide accurate nuclear shieldings.61 The convergence of 13C nuclear shieldings in C60 obtained with pcS-n (n = 0, 1, 2, 3) can be seen in Fig. S1 (ESI†). Table S1 (ESI†) lists the 13C chemical shifts for C70 calculated using different pcS-n (n = 0, 1, 2) basis sets. It is apparent that the IGLO-n basis sets provided faster convergence than pcS-n.
Performance of various density functionals
The results obtained using the selected DFT functionals are summarized in Table 2, where the calculated 13C shifts as well as their root-mean-square deviations (RMSD) from the experimental values are collected. The results in Table 2 are ordered according to the RMSD. Typically, the DFT calculations overestimate the experimental 13C NMR chemical shift by several ppm. Obviously, the effects of solvent and temperature are not included at this stage, and we can compare only the main trends exhibited by the functionals.| Functional | Ca | Cb | Cc | Cd | Ce | RMSDa (RMSDb) |
|---|---|---|---|---|---|---|
| a Ref. 13. b This work. c Values using B3LYP/6-31G* taken from ref. 10. Calculations were performed for BP86/def-TZVP optimized geometry and with the IGLO-III basis set. | ||||||
| wB97xd | 150.6 | 147.3 | 147.9 | 145.8 | 132.3 | 0.4 (0.4) |
| CAMB3LYP | 151.8 | 148.0 | 149.0 | 146.7 | 132.5 | 0.6 (0.7) |
| M06L | 152.8 | 148.0 | 149.7 | 146.6 | 130.8 | 0.7 (0.7) |
| OPBE | 152.9 | 148.4 | 149.9 | 147.1 | 129.9 | 0.8 (0.8) |
| BHandHLYP | 152.7 | 148.6 | 149.9 | 147.0 | 132.7 | 0.8 (0.9) |
| BHandH | 152.8 | 148.8 | 150.1 | 147.1 | 132.1 | 0.9 (0.9) |
| O3LYP | 153.2 | 148.7 | 150.3 | 147.4 | 131.1 | 0.9 (1.0) |
| HCTH | 153.3 | 148.6 | 150.3 | 147.4 | 130.5 | 0.9 (1.0) |
| OLYP | 153.5 | 148.8 | 150.4 | 147.5 | 130.5 | 1.0 (1.0) |
| B98 | 153.6 | 149.2 | 150.7 | 147.8 | 131.6 | 1.1 (1.1) |
| B1LYP | 153.7 | 149.2 | 150.7 | 147.8 | 132.0 | 1.1 (1.2) |
| X3LYP | 153.8 | 149.3 | 150.8 | 147.9 | 131.8 | 1.1 (1.2) |
| B3LYP | 153.8 | 149.3 | 150.9 | 147.9 | 131.7 | 1.1 (1.2) |
| PBEPBE | 154.2 | 149.5 | 151.1 | 148.0 | 130.4 | 1.2 (1.3) |
| BLYP | 154.8 | 150.0 | 151.5 | 148.5 | 131.0 | 1.4 (1.5) |
| SV5LYP | 154.8 | 150.1 | 151.6 | 148.5 | 129.7 | 1.4 (1.5) |
| M06HF | 155.5 | 152.3 | 151.0 | 148.5 | 132.7 | 1.7 (1.8) |
| BP86 | 155.6 | 150.8 | 152.4 | 149.4 | 131.9 | 1.7 (1.8) |
| Exp.a | 150.8 | 147.8 | 148.3 | 144.4 | 130.8 | — |
| Exp.b | 150.3 | 147.1 | 147.8 | 145.0 | 130.5 | — |
| B3LYPc | 151.2 | 147.0 | 148.2 | 146.3 | 132.1 | 0.5 (0.5) |
Nevertheless, for the gas-phase C70 molecule at rest it appears that the long-range corrected functionals CAM-B3LYP and wB97xd as well as the hybrid functionals with 50% of exact-exchange admixture BHandH and BHandHLYP perform the best, while the non-hybrid GGA functionals give a worse agreement with the experiment. The OPBE functional, provides results of the same accuracy as BHandHLYP with much smaller CPU cost. The good performance of the OPBE62,63 as well as “half-and-half” functionals for NMR parameters has been noticed elsewhere.58,59,64–67 A good performance is provided also by the M06L pure DFT functional of Truhlar and Zhao.33 Combining different exchange functionals with the LYP correlation along the B1LYP, B3LYP, X3LYP, and O3LYP series shows rather marginal dependence of the performance on the exchange functional. Increasing the exact exchange admixture in the functional generally lowers the calculated 13C chemical shifts, bringing them closer to the experimental ones, as seen in Fig. 2. A decrease of the RMSD with increasing exact-exchange admixture is observed for relative carbon shifts (C signals relative to Ce) in Fig. 2. Noticeably, if one looks at relative 13C NMR chemical shifts in Table S2 (ESI†) instead of the absolute ones in Table 2 the relative performance is somewhat altered especially for larger RMDS values.
 | ||
| Fig. 2 The effect of increasing exact-exchange admixture in DFT functionals on calculated 13C chemical shifts. RMSD (solid line) stands for root mean square deviations from the experimental values.13 RMSDr (dashed line) is calculated for relative 13C shifts (C–Ce). | ||
We did not include the multiplicative Kohn–Sham methods68 denoted also as the optimized-effective potential (OEP) methods, often leading to improvements in calculations of magnetic resonance properties.69–75However, less significant improvement for carbon only containing compounds was observed.76
If the larger IGLO-IV basis set was used in the calculations shown in Table 2 (not affordable presently), most RMSDs should improve, as the DFT calculated 13C chemical shifts would lower by about 0.1–0.2 ppm, mostly towards the experimental values. We do not expect this rather small basis set effect to alter the performance of the DFT functionals, as shown in Table 2.
The solvent effect
As seen in Table 3, the inclusion of the solvent within the CPCM implicit solvent model moderately decreases the calculated 13C shifts by 0.5–1.0 ppm towards the experimental values. Note that the new experiment performed in this work in ODCB solvent (ε = 9.93) provided almost identical results to the previous experiments done in 1,1,2,2-tetrachloroethane (see Table 1). This suggests a limited dependence of the 13C NMR chemical shifts on the kind of solvent for this non-polar molecule. A closer look at Table 3 reveals that the BHandH and OLYP functionals decrease calculated shifts towards experimental results (RMSD drop of ∼0.1 ppm), while including the effects of solvent slightly increases the RMSD error at the wB97xd level, from 0.4 to 0.5.| Exp. | BHandH | OLYP | wB97xd | ||||
|---|---|---|---|---|---|---|---|
| Vac. | Solv. | Vac. | Solv. | Vac. | Solv. | ||
| a The BP86/def-TZVP or BP86/def-TZVP/CPCM geometry was used. Implicit solvent with parameters for 1,1,2-trichloroethane and the IGLO-III basis set for shielding calculations were employed. RMSD is the root-mean-square deviation from experimental values in ref. 13. RMSDr is calculated for relative chemical shifts (C–Ce). | |||||||
| Ca | 150.8 | 152.8 | 152.3 | 153.5 | 153.2 | 150.6 | 150.2 |
| Cb | 147.8 | 148.8 | 148.2 | 148.8 | 148.4 | 147.3 | 146.7 |
| Cc | 148.3 | 150.1 | 149.6 | 150.4 | 150.1 | 147.9 | 147.5 |
| Cd | 144.4 | 147.1 | 146.9 | 147.5 | 147.4 | 145.8 | 145.5 |
| Ce | 130.8 | 132.1 | 131.8 | 130.5 | 130.3 | 132.3 | 132.0 |
| RMSD | 0.8 | 0.7 | 1.0 | 0.9 | 0.4 | 0.5 | |
| RMSDr | 0.4 | 0.4 | 1.3 | 1.3 | 0.8 | 0.9 | |
Equilibrium geometry effect
The effect of the equilibrium geometry on the calculated 13C shifts for C70 is illustrated in Table 4. While improving the basis set from def-TZVP towards def2-QZVP, the shift decreases towards the experiment in a uniform way, by about 1 ppm, when comparing the def-TZVP and def2-QZVP structures.| Structure | Ca | Cb | Cc | Cd | Ce | RMSD (RMSDr) |
|---|---|---|---|---|---|---|
| a The BP86 functional was used. b The def2-TZVP basis set was used. RMSD is the root-mean-square deviation from experimental values ref. 13, RMSDr is calculated for relative chemical shifts (C–Ce). | ||||||
| def-SVPa | 152.2 | 148.2 | 149.5 | 146.5 | 131.6 | 0.6 (0.4) |
| def-TZVPa | 152.8 | 148.8 | 150.1 | 147.1 | 132.1 | 0.8 (0.4) |
| def2-TZVPa | 151.9 | 147.9 | 149.3 | 146.5 | 131.2 | 0.5 (0.5) |
| def2-QZVPa | 151.9 | 147.8 | 149.2 | 146.4 | 131.2 | 0.5 (0.5) |
| BP86b | 152.8 | 148.8 | 150.1 | 147.1 | 132.1 | 0.8 (0.4) |
| B3LYPb | 151.9 | 147.8 | 149.0 | 146.5 | 131.2 | 0.5 (0.5) |
| B97db | 155.1 | 151.0 | 152.3 | 149.7 | 134.6 | 1.9 (0.4) |
| Exp. | 150.8 | 147.8 | 148.3 | 144.4 | 130.8 | |
Changing the DFT functional used for the optimization leads to substantially larger shift changes (obtained at the BHandHLYP/IGLO-III level), and they can both improve or worsen the results, as seen in Table 4. The B3LYP structure provides chemical shifts closer to experiment, while the B97d structure largely overestimates the shifts. If relative chemical shifts are compared to experimental results in Table 4via RMSDr (obtained from C signals relative to Ce), the B3LYP functional provides worse agreement than BP86 and B97d.
The seemingly good performance of, e.g. the B3LYP/def2-TZVP structure as compared to our default level BP86/def-TZVP is presumably due to error cancellation, as seen in Fig. 3, where the C–C distances in C60 calculated at different levels are compared to the experimental ones. The B3LYP/def2-TZVP or BHandHLYP/def2-TZVP levels provide worse calculated C–C distances than our default level for structure optimization BP86/def-TZVP. The BP86/def-TZVP level appears to provide reliable molecular structures also for endohedral helium fullerenes.58,59
 | ||
| Fig. 3 Calculated bond lengths in optimized C60 obtained using the BP86 functional and different basis sets (on the right) and density functionals in combination with the def2-TZVP basis set (on the left). Experimental results are obtained from ref. 77. The C–C bonds are displayed on the top. | ||
The molecular motions and 13C chemical shifts
The results summarizing different strategies for accounting of the temperature and dynamics are summarized in Table 5, as corrections to individual carbon nuclear magnetic shieldings, i.e. differences between dynamically/vibrationally averaged and optimized-structure nuclear shieldings. Averaged distances and spatial fluctuations of carbon atoms derived from the simulations are detailed in Table S3 (ESI†).| Method | Ca | Cb | Cc | Cd | Ce | C60 |
|---|---|---|---|---|---|---|
| a Potential HF/6-31G//level for NMR BP86/IGLO-II. b Potential MM3//level for NMR BP86/IGLO-II. c Potential BP86/def-SVP//level for NMR BHandHLYP/IGLO-III. | ||||||
| PT2a | −4.6 | −4.5 | −4.6 | −4.5 | −4.5 | — |
| VCIa | −5.0 | −5.0 | −5.1 | −5.5 | −6.3 | — |
| TCSa | −7.3 | −7.2 | −7.3 | −7.9 | −9.2 | — |
| MDb | −6.0 | −3.8 | −5.0 | −5.7 | 2.1 | −3.4 |
| FPMDc | −1.4 | −2.0 | −2.3 | −2.4 | −5.8 | −1.4 |
| FPMD/COSMOc | −1.3 | −1.2 | −1.2 | −1.5 | −1.7 | −1.2 |
The changes caused by the molecular motions for the gas-phase C70 molecule can be assigned as deshielding and are relatively large, up to ca. −9 ppm, very much depending on the method. The positive (shielding) contribution result for Ce in the MD line in Table 5 may be an artifact caused by the inaccurate force field. The vibrational averaging in PT2 approximation gives nearly uniform deshielding of about −4.5 ppm for all carbon types, while the TCS, classical MD, and FPMD approaches show relatively more variable effects. The corrections for carbon Ce are significantly different from the other carbon types. The geometric fluctuations of atoms Ca–Ce around the equilibrium position are rather uniform (Table S4, ESI†), hence we infer that the differential dynamical contributions for Ca–Ce in FPMD originate from the fluctuations in the electronic structure.
For the C60 reference, dynamical correction to 13C nuclear magnetic shielding about −3 to −2 ppm was obtained using different methods comparable with that of −2.5 ppm mentioned previously.78 If anomalous contribution for atom Ce in C70 is excluded then the “dynamical” contribution for C60 represents approximately 70% of average C70 values (MD, FPMD). The vibrational (PT2, VCI, TCS) contributions to the 13C NMR chemical shifts in C70 are likely to be similar as in C60. Therefore, the usage of C60 as the secondary reference will partly cancel the dynamical corrections.
If the best DFT results in Table 2 are added to the dynamical corrections in Table 5 (taken with opposite sign to turn the nuclear magnetic shielding into the chemical shift), the agreement with the experiment is not improved. However, when the FPMD simulations are performed in the solvent, the dynamical corrections (Table 5) for the 13C nuclear magnetic shielding are still deshielding but now considerably smaller in absolute size and more uniform (−1.2 to −1.7 ppm) than those obtained for FPMD simulation in vacuo. A detailed look reveals that the solvent damps the molecular motions. This is documented in Table S2 (ESI†) on the average deviations from equilibrium positions.
The different contributions to the total 13C NMR chemical shifts in C70 discussed above are summarized in Fig. 4. The inclusion of solvent and basis set correction decreases the gas phase shifts and thus improve the agreement with experiment.
 | ||
| Fig. 4 Importance of various effects on calculated 13C chemical shifts in C70. BHandHLYP stands for shifts obtained by the vacuum BHandHLYP/IGLO-III calculation, “solvent” denotes the BHandHLYP/IGLO-III/CPCM model, and the other bars are related to the involvement of vacuum and solvent FPMD dynamics. All data are corrected for the estimated basis set error calculated from difference between the IGLO-IV and IGLO-III results in Table 1 (all results comprise the previous enhancements, e.g. the “dynamic” = BHandHLYP shifts in vacuum + solvent contribution + basis set correction + dynamic contribution). The experiment is taken from ref. 13. | ||
The contributions due to molecular motions (FPMD is shown as example in Fig. 4) are mostly positive, and shift the values away from the experimental ones. If the FPMD simulations could be run longer, an improvement of a couple of tenths of ppm could be expected due to somewhat large statistical error (mean error of the average, see the Methods section) obtained, ±0.35 ppm for the dynamical corrections to the 13C chemical shift. The total calculated 13C shifts corrected for the basis set error and dynamical solvent are again closer to the experimental ones. However, the inclusion of the dynamical corrections including solvent does not bring any significant improvement over the static molecule in vacuo results, because they are small in size, relatively uniform, and largely cancelled by the dynamical effect of the reference C60 compound.
The temperature dependence
Vibrational averaging obtained using the TCS method also enables the estimation of the temperature dependence of the carbon chemical shifts in C70. Results for 273 K and 373 K calculated using two different computational levels are shown in Table S4 (ESI†). The calculated and experimental changes (related to value for carbon Ce) are shown in Fig. 5. The temperature dependence appears to be relatively uniform for all carbons at both levels, ∼0.6–0.9 ppm per 100 K. Experimental dependence can be seen in Fig. S2 (ESI†). The correlation between experimental and theoretical results is clear. However, the observed change is about 5 times smaller than in theory. This can be partially attributed to the missing solvent effects in the TCS calculations, which, however, could not be included due to the computational time limits. | ||
| Fig. 5 Temperature effect on NMR shifts in C70. Plot shows comparison of experimental and theoretical (BP86/IGLO-II//HF/6-31G) differences in relative shieldings caused by temperature (Δ = 100 K). The shift is referenced to Ce. | ||
Conclusions
To estimate the accuracy of available theoretical procedures, 13C NMR chemical shifts in the C70 fullerene were modelled using various density functionals and basis sets. The effects of solvent, dynamics, and temperature on the 13C shifts in the theoretical simulations were discussed. Reasonable 13C chemical shift values within several ppm from the experimental results could be obtained already by the usual calculations of a gas-phase molecule at rest. With hybrid or long-range corrected functionals the calculated chemical shifts were overestimated by about 2 ppm. The largest errors, up to 5 ppm, were found for the GGA functionals, such as BP86. The basis set effects on the 13C shifts calculated for the IGLO-II to IGLO-IV series are moderate; the errors using the IGLO-III basis set are estimated within 0.4 ppm of the basis set limit, or 0.2 ppm from IGLO-IV results. Improving the basis set size in the structure optimization resulted in changes a few tenths of ppm on the 13C NMR chemical shifts, whereas changing the functional had a larger impact of several ppm.Inclusion of the implicit solvent effects caused a decrease of the calculated 13C chemical shifts towards the experimental values by about 1 ppm. The effect of molecular motions on the 13C NMR chemical shift in gas-phase C70 caused relatively large deshielding contributions of about 2–9 ppm, depending on the method used for averaging. These changes were strongly damped if the solvent was considered in simulation, leading to a deshielding contribution of about 1.2–1.7 ppm only. For the chemical shifts, the changes were largely cancelled by the motional contributions from the C60 reference.
Acknowledgements
This work was supported by the Grant Agency of the Czech Republic (203/09/0237, 13-03978S, P208/10/P356, P208/11/0105), Ministry of Education (LH11033), and Czech Academy of Sciences (M200551205).Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- L. Dunsch and S. Yang, Phys. Chem. Chem. Phys., 2007, 9, 3067–3081 RSC.
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- P. W. Fowler and D. E. Manolopoulos, An Atlas of Fullerenes, Dover Publications, Dover, 2006 Search PubMed.
- G. Y. Sun and M. Kertesz, J. Phys. Chem. A, 2002, 106, 6381–6386 CrossRef CAS.
- G. Sun and M. Kertesz, Chem. Phys., 2002, 276, 107–114 CrossRef CAS.
- G. Sun and M. Kertesz, J. Phys. Chem. A, 2001, 105, 5212–5220 CrossRef CAS.
- G. Sun and M. Kertesz, New J. Chem., 2000, 24, 741–743 RSC.
- G. Sun and M. Kertesz, J. Phys. Chem. A, 2001, 105, 5468–5472 CrossRef CAS.
- G. Sun and M. Kertesz, J. Phys. Chem. A, 2000, 104, 7398–7403 CrossRef CAS.
- G. Sun and M. Kertesz, Chem. Phys. Lett., 2000, 328, 387–395 CrossRef CAS.
- T. Heine, in Calculation of NMR and EPR Parameters: Theory and Applications, ed. M. Kaupp, M. Bühl and V. G. Malkin, Wiley-VCH, Weinheim, 2004, pp. 409–419 Search PubMed.
- F. Diederich and R. L. Whetten, Acc. Chem. Res., 1992, 25, 119–126 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- B. C. Mort and J. Autschbach, J. Phys. Chem. A, 2005, 109, 8617–8623 CrossRef CAS.
- M. Dračínský, J. Kaminský and P. Bouř, J. Chem. Phys., 2009, 130, 094106 CrossRef.
- R. Ahlrichs, M. Bar, H.-P. Baron, R. Bauernschmitt, S. Bocker, M. Ehrig, K. Eichkorn, S. Elliot, F. Furche, F. Haase, M. Haser, H. Horn, C. Huber, U. Huniar, M. Kattannek, C. Kolmel, M. Koolwitz, K. May, C. Ochsenfeld, H. Ohm, A. Schafer, U. Schneider, O. Treutler, M. von Arnim, F. Weigend, P. Weis and H. Weiss, TURBOMOLE V6.3.1 2011, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Wallingford CT, 2009.
- J. W. Ponder, Tinker – Software Tools for Molecular Design, St. Louis, 2009 Search PubMed.
- P. Ren and J. W. Ponder, J. Phys. Chem. B, 2003, 107, 5933–5947 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. A. Hamprecht, A. Cohen, D. J. Tozer and N. C. Handy, J. Chem. Phys., 1998, 109, 6264–6271 CrossRef CAS.
- C. Lee, W. Yang and R. G. Paar, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- N. C. Handy and A. J. Cohen, Mol. Phys., 2001, 99, 403–412 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. C. Slater, The Self-Consistent Field for Molecular and Solids, Quantum Theory of Molecular and Solids, McGraw-Hill, New York, 1974, vol. 4 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194118 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 104, 1040–1046 CrossRef.
- A. J. Cohen and N. C. Handy, Mol. Phys., 2001, 99, 607–615 CrossRef CAS.
- X. Xu and W. A. Goddard III, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2673–2677 CrossRef CAS.
- H. L. Schmider and A. D. Becke, J. Chem. Phys., 1998, 108, 9624–9631 CrossRef CAS.
- H. Zhao and D. G. Truhlar, J. Phys. Chem., 2006, 110, 5121–5129 CrossRef.
- T. Yanai, D. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- W. Kutzelnigg, U. Fleischer and M. Schindler, The IGLO-Method: Ab Initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities, Springer-Verlag, Heidelberg, 1990 Search PubMed.
- S. Huzinaga, Approximate Atomic Functions, University of Alberta, Edmonton, 1971.
- W. Kutzelnigg, U. Fleischer and M. Schindler, NMR – Basic Principles and Progress, Springer-Verlag, Heidelberg, 1990 Search PubMed.
- F. Jensen, J. Chem. Theory Comput., 2008, 4, 719–727 CrossRef CAS.
- A. G. Avent, D. Dubois, A. Penicaud and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1997, 1907 RSC.
- D. Papoušek and M. R. Aliev, Molecular Vibrational/Rotational Spectra, Academia, Prague, 1982 Search PubMed.
- P. Daněček and P. Bouř, J. Comput. Chem., 2007, 28, 1617–1624 CrossRef.
- P. Bouř, S4 program for anharmonic molecular vibrations, Academy of Sciences, Prague, 2012 Search PubMed.
- J. A. Pople, H. B. Schlegel, R. Krishnan, D. J. DeFrees, J. S. Binkley, M. J. Frisch, R. A. Whiteside, R. F. Hout and W. J. Hehre, Int. J. Quantum Chem., Quantum Biol. Symp., 1981, 15, 269 CAS.
- R. F. Hout, B. A. Levi and W. J. Hehre, J. Comput. Chem., 1982, 3, 234 CrossRef CAS.
- P. A. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502–16513 CrossRef.
- G. Rauhut and P. Pulay, J. Phys. Chem., 1995, 99, 3093 CrossRef CAS.
- B. G. Johnson, P. M. W. Gill and J. A. Pople, J. Chem. Phys., 1993, 98, 5612 CrossRef CAS.
- R. H. Hertwig and W. Koch, J. Comput. Chem., 1995, 16, 576 CrossRef CAS.
- M. Dračínský and P. Bouř, J. Comput. Chem., 2012, 33, 1080–1089 CrossRef.
- B. C. Mort and J. Autschbach, J. Phys. Chem., 2005, 109, 8617 CrossRef CAS.
- N. L. Allinger, Y. H. Yuh and J.-H. Lii, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- F. Jensen, Introduction to Computational Chemistry, Willey-VCH, Weinheim, 2nd edn, 2007 Search PubMed.
- M. Straka and J. Vaara, J. Phys. Chem. A, 2006, 110, 12338–12341 CrossRef CAS.
- P. Štěpánek, P. Bouř and M. Straka, Chem. Phys. Lett., 2010, 500, 54–58 CrossRef.
- A. A. Auer, J. Gauss and J. F. Stanton, J. Chem. Phys., 2003, 118, 10407 CrossRef CAS.
- T. Kupka, M. Stachow, M. Nieradka, J. Kaminský, T. Pluta and S. P. A. Sauer, Magn. Reson. Chem., 2011, 49, 231–236 CrossRef CAS.
- A. Wu, Y. Zhang, X. Xu and Y. Yan, J. Comput. Chem., 2007, 28, 2431–2442 CrossRef CAS.
- Y. Zhang, A. Wu, X. Xu and Y. Yan, Chem. Phys. Lett., 2006, 421, 383–388 CrossRef CAS.
- T. Kupka, M. Stachow, M. Nieradka, J. Kaminský and T. Pluta, J. Chem. Theory Comput., 2010, 6, 1580–1589 CrossRef CAS.
- T. Kupka, M. Nieradka, M. Stachow, T. Pluta, P. Nowak, H. Kjaer, J. Kongsted and J. Kaminský, J. Phys. Chem. A, 2012, 116, 3728–3738 CrossRef CAS.
- M. Straka, P. Lantto and J. Vaara, J. Phys. Chem. A, 2008, 112, 2658–2668 CrossRef CAS.
- S. Standara, P. Kulhánek, P. Bouř, R. Marek, J. Horníček and M. Straka, Theor. Chem. Acc., 2011, 129, 677–684 CrossRef CAS.
- Q. Zhao, R. C. Morrison and R. G. Parr, Phys. Rev. A: At., Mol., Opt. Phys., 1994, 50, 2138 CrossRef CAS.
- P. J. Wilson and D. J. Tozer, Chem. Phys. Lett., 2001, 337, 341–348 CrossRef CAS.
- M. J. Allen, T. W. Keal and D. J. Tozer, Chem. Phys. Lett., 2003, 380, 70–77 CrossRef CAS.
- A. M. Teale and D. J. Tozer, Chem. Phys. Lett., 2004, 383, 109–114 CrossRef CAS.
- A. V. Arbuznikov and M. Kaupp, Chem. Phys. Lett., 2004, 386, 8–16 CrossRef CAS.
- A. V. Arbuznikov and M. Kaupp, Chem. Phys. Lett., 2004, 391, 16–21 CrossRef CAS.
- A. J. Cohen, Q. Wu and W. Yang, Chem. Phys. Lett., 2004, 399, 84–88 CrossRef CAS.
- P. J. Wilson and D. J. Tozer, J. Mol. Struct., 2002, 602–603, 191–197 CrossRef CAS.
- A. M. Teale, O. B. Lutnaes, T. Helgaker, D. J. Tozer and J. Gauss, J. Chem. Phys., 2013, 138, 024111 CrossRef.
- K. Hedberg, L. Hedberg, D. S. Bethune, C. A. Brown, H. C. Dorn, R. D. Johnson and V. M. De, Science, 1991, 254, 410–412 CAS.
- T. Heine, K. Vietze and G. Seifert, Magn. Reson. Chem., 2004, 42, S199–S201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Modified MM3 fullerene parameters, performance of DFT functionals for 13C chemical shifts, calculated data for TCS, the effect of temperature on the experimental 13C NMR signal. See DOI: 10.1039/c3cp50657f |
| This journal is © the Owner Societies 2013 |