 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linking electronic and molecular structure: insight into aqueous chloride solvation†
Ling
Ge
*a,
Leonardo
Bernasconi
b and
Patricia
Hunt
*a
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: l.ge@imperial.ac.uk
bSTFC Rutherford Appleton Laboratory, Didcot, Oxfordshire OX11 0QX, UK
First published on 31st May 2013
Abstract
Aqueous chloride solutions are ubiquitous and diverse; systems include sea water, atmospheric droplets, geological processes and biological organisms. However, despite considerable effort, a complete microscopic model of the hydration shell, and local electronic structure of the aqueous chloride ion and its dynamics has not been established. In this work we employ ab initio molecular dynamics to study an aqueous chloride solution. In particular, local solvation events and the electronic structure around the chloride ion are interrogated. We employ the Effective Molecular Orbital (EMO) method which partitions the electronic structure into solute and solvent components while maintaining a rigorous quantum mechanical description of both. Movement of the chloride highest occupied molecular orbital (HOMO) energy within the valence band of water is revealed. The chloride ion has little impact on the average water electronic structure, however, locally the electronic effect of the chloride ion is significant. With the Hofmeister series in mind we find that the electronic effect of the chloride ion extends beyond the first solvation shell, but not beyond the edge of the second solvation shell. The chloride ion sits near the centre of the Hofmeister series because of an essential degeneracy between water–water and water–Cl H-bonding and because of a strong similarity in the water and chloride electronic structure. The chloride ion prefers to be symmetrically solvated by six H-bonding water molecules, however, the chloride HOMO energy and the coordination number oscillate in response to local fluctuations driven by the dynamics of the bulk water. A combined structural and electronic analysis has led to a distinction between two types of water molecule within the first solvation shell, those that H-bond to the chloride ion, and those that remain local (i.e. within the first solvation shell) but which H-bond to other water molecules. There are indications that these exhibit different dynamics with respect to residence times and rotational vs. translational motion.
1 Introduction
Chloride solutions are of key importance in atmospheric chemistry, geological processes and biochemical systems. Salt water covers 2/3 of the earth’s surface, atmospheric aerosols impact on the radiative balance of the earth, and water forms the main constituent of most living organisms. The generation of clean non-saline water is a key challenge for the coming century, and water from anthropogenic or natural environmental (oceanic, land based, and atmospheric) sources impacts on climate change and global warming. In all of these processes the presence of salts, and principally aqueous chloride ions, is ubiquitous.Cl anions can enter the atmosphere from the sea surface. Airborne aqueous sea-salt aerosols are a global source of molecular chlorine in the troposphere.1–3 Once in the atmosphere, Cl anions impact on the ozone cycle.4 They also act as nucleation sites for aerosols which perturb the radiation balance of the earth.5 A significant source of uncertainty in the current understanding of global climate change is atmospheric aerosol radiative forcing.6
Natural water sources contain Cl anions which play an important role in oceanographic and geological processes. For example, small variations in sea surface salinity can have dramatic effects on the water cycle and ocean circulation.7 Changing environmental conditions can alter processes such as the diffusive transport of saltwater into freshwater sediments. This in turn effects the present and historical character of sediments in lakes and lagoons.8 With reducing fresh water sources, the processing of saline water has become of increasing importance. The models used to understand Cl anion solute transport require knowledge of the hydration environment and reactivity of the Cl anion in aqueous solution.
In biological systems, the ability of salts to induce or retard the precipitation of proteins and non-polar (hydrocarbon) solutes from an aqueous solution is important, and follows a trend known as the Hofmeister series.9–13 Similar trends are found for surface tension, chromatographic selectivity, protein denaturation temperatures, protein stability, protein–protein interactions, protein crystallisation, enzyme activity, and colloid stability.9,10,13 Properties such as the Jones–Dole B coefficient of viscosity, the permeation of ions through membranes, measurements on pH buffers, zeta and membrane potentials, colloidal force measurements, ion pumps, neutron scattering and direct force measurements are all interpreted in terms of classical models including ion (Hofmeister) and hydration sphere effects.9,14,15
Traditionally, the Hofmeister series has been related to the effect of an ion on the (H-bonding) structure of liquid water. Structure makers (kosmotropes) decrease the solubility of non-polar molecules (“salting out”). Structure breakers (chaotropes) enhance the solubility of non-polar molecules (“salting in”).16 The increased solubility of non-polar molecules is due to a reduction in the negative entropy (or enhanced disorder) in the surrounding solvent, structure breakers facilitate this process. Structure makers are assumed to enhance the tetrahedral network of water making it more icelike.
Small anions with a higher charge density have a tight first solvation shell, they form H-bonds to local water molecules and are structure makers. Small ions are also repelled from an air–water interface. Larger but more diffuse anions without a strong solvation shell are structure breakers. Large ions tend to gather at the air–water interface. This study focuses on the Cl anion, which sits near the middle of the Hofmeister series for anions: SO42− > F− > CH3COO− > Cl− > Br− > I− > SCN−. The Cl anion is generally considered a weak structure breaker,10,14 and is known to be neutral with respect to a propensity to collect at the water–air boundary.17,18 The Cl anion is small enough to be hydrated, but it is also large enough to be polarizable.
Experimental and theoretical results from the literature (outlined shortly) reveal a rather inhomogeneous picture of the hydrated chloride ion solvation shell structure. Moreover, there is little information about the underlying electronic structure which drives the dynamics associated with solvation. In this paper, we link the electronic and molecular structure and provide insight into aqueous chloride solvation.
The effect of aqueous solvated ions beyond the first or the second solvation shell is less clear, particularly as this applies to H-bonding between water molecules, and this topic has generated significant debate in the recent literature.13,16,19 There is physical and simulation based evidence for both the traditional view of long range ion effects,14,20–24 and a more recent proposal that ion effects are purely local.15,25–33 There is also evidence that hydration effects beyond the first solvation shell are dependent on the particular cation–anion combination and on the concentration.20,27,28,34 Moreover, some studies show effects, which are minor at a long range but significant at a short range.20,35 It has also been suggested that the effects do not relate to the structuring of water, but that ions interact directly with the protein.13,36 How a tight solvation sphere enhances the long range structure in water, or how a large polarizable anion disrupts the long range structure in water is significantly more tenuous and not easy to rationalize, particularly in light of the dynamic nature of liquid water. It is a significant issue that the Hofmeister series cannot be rationalised by reference to a molecular level picture.24,37 It has also been suggested that the terms “structure maker” and “structure breaker” are not supported at the molecular scale.20
Simulations have indicated that fixed charge models are not sufficient to describe ions in aqueous solution, and the ability to induce a dipole centered on the ion is important.16,17 However, it has also been found that Hofmeister related phenomena can also be recovered via other routes, such as increasing the size of the anion, using optimised non-polarizable force fields, including electrostatic damping, or by considering the air/water interfacial shape.38–41
Thus, there is clear motivation for investigating the solvation of small ions, and the chloride ion in particular. It is important to understand solvation at the molecular level because this information impacts on our understanding and modelling of atmospheric, land and sea based water bodies, geological processes and biochemical systems. These molecular scale phenomena are ultimately linked to key current global challenges; climate change prediction and clean water generation.
A variety of experimental methods have been used to investigate the hydration structure of chloride ions in water, these include neutron and X-ray diffraction techniques,20,21,23,35,42,43 X-ray absorption spectroscopy (EXAFS and XANES),44 gel sieving chromatography,45 Raman and IR spectroscopy,30,46 mass spectroscopy47,48 and photoelectron spectroscopy.49 More recently the development of new spectroscopic methods, femtosecond infrared pump–probe IR spectroscopy and terahertz dielectric relaxation spectroscopy, has provided additional details.10,25,50–52 The optical Kerr effect and dielectric relaxation spectroscopy have also offered insight.53 However, experimentally derived coordination numbers are significantly scattered, ranging from 3 to 8.20,21,44,46,49,54 Thus the number of water molecules surrounding the chloride ion in the first solvation shell is still unclear.
A range of theoretical methods, from molecular dynamics (MD) simulations through ab initio MD and full quantum chemical calculations, have been applied to the water–chloride interaction and Cl anion solvation. Difficulties in determining the structure of the solvation environment from MD studies arise because of the sensitivity of results to the details of water–Cl and water–water interaction potentials.17,55–60 Differently parametrized potentials, with and without a treatment of molecular polarizability, give different structural properties.24,61,62 For example, simulations of small hydrated clusters have demonstrated that changing the polarizability of the “chloride” ion determines whether the whole cluster is symmetric or polarised.63 In contrast, simulations of larger clusters (20–255 water molecules) have indicated that the water (and not ions) polarisability is important.61,64 Non-polarizable potentials tend to predict an interior location for the chloride anion, while polarizable models predict a surface location for the anion.65 More recently it has been suggested that the degree of polarisation induced in classical models may be too strong.66Ab initio MD naturally recovers polarisation for both the ion and water; studies have been carried out on aqueous chloride solutions,67–69 and chloride water clusters.65,70 In both cases an asymmetric solvation structure is found. Quantum mechanics/molecular mechanics simulations (employing a HF QM description) of a Cl anion in water have also been carried out.71 Uncertainty in the coordination number is evinced from theoretical studies, with variations between 5.1 and 8.4 depending on the simulation techniques adopted.56,60,67,68,71–77 Thus, despite extensive efforts, a complete microscopic model of the hydration shell of the aqueous chloride ion and its dynamics is still being debated.
Thus, the solvation of Cl anions in an aqueous environment has been the subject of a substantial amount of experimental and theoretical work. Nevertheless, despite concerted efforts to unravel the effect of ions on the structure, dynamics and electronic features of water, many questions remain open. How chloride ions are accommodated in the solvent hydrogen bonding network, and how they influence the solvent structure, is not well understood. Moreover, there is little information on the intimate connection between the electronic structure of the solute chloride ion and aqueous solvent.
In this work we employ ab initio MD to study an aqueous chloride solution and in particular focus on the local solvation structure around the Cl anion. In contrast to previous ab initio studies, we concentrate on the nature of the electronic interactions between the solute Cl anion and the solvent, which is interpreted on the basis of the Effective Molecular Orbital (EMO) method.78 A combination of the EMO method and a more traditional structural analysis is then used to examine the relationship between the coordination environment and the electronic structure of the Cl anion. Moreover, we investigate the extent to which the Cl anion influences water molecules beyond the first solvation shell.
The paper is organised as follows. In Section 2 the computational details of the MD simulations are presented. In the Results and discussion (Section 3) radial distribution functions and coordination numbers are discussed and benchmarked. The results of the EMO analysis are presented, the extent of electronic coupling between the chloride ion and water molecules is analysed, the local solvation environment is interrogated, the dipole moments of the system reported and rare events which involve water molecules entering or leaving the first solvation shell are analysed. Results are summarised in the Conclusions (Section 4).
2 Computational details
Ab initio MD calculations of a Cl anion solvated in liquid water have been performed using the CPMD program.79,80 The plane wave-pseudopotential implementation of the Kohn–Sham method used in this code has been validated in previous studies of aqueous systems.78,81–83 Similar to the majority of these simulations the BLYP functional has been employed.84,85 Norm conserving pseudo potentials have been employed and expanded in a plane wave basis up to an energy cutoff of 70 Ry at the Γ point.The system consists of 63 water molecules and one chloride ion in a simple cubic supercell of dimension 12.42 Å corresponding to the experimental density under ambient conditions with chloride ion concentration (0.88 mol L−1). The starting configuration was obtained from an equilibrated classical MD simulation using an extended simple point charge (SPC/E) model for water, in combination with a chloride–water potential.60,86 The ab initio trajectory was then equilibrated to an average temperature of 298 K using CPMD for 2.76 ps. A fictitious mass of 600 a.u. and a time step of 5 a.u. were used in the integration of the equations of motion. The simulation temperature was controlled through the equilibration phase via a Nosé–Hoover thermostat with a frequency of 1100 cm−1.87 In a real system the Cl anion will always be present with a neutralising cation, however it has been found experimentally that the Cl anion solvation structure is predominantly independent of the nature of the counter ion.20,21,35,46,52 In these simulations the counter ion has been replaced by a positive neutralising background charge. For the electronic analysis 80 structures were collected over the post-equilibration phase, of 28.8 ps at equal intervals of 0.36 ps.
The EMO method interprets the time evolution of the electronic and molecular structure of both solute and solvent.78 This analysis is based on a rigorous quantum mechanical description of both the solute and solvent, i.e. it does not rely on partitioning of the system into quantum-mechanical (solute) and classical (solvent) components. A brief description of the method can be found in the Fig. S1 and S2 (ESI†). The EMO analysis determines an effective energy α for each EMO and β which represents the electronic coupling between orbitals on different molecules. Thermal broadening can be obtained as the standard deviation in the energy of the EMOs, Δα.
The EMO analysis has been successfully applied to pure liquid water.78 An isolated water molecule has C2v symmetry and valence MOs 2a1, 1b1, 3a1 and 1b2 (Fig. S3, ESI†). A liquid environment is disordered and there is no symmetry, however, the EMOs strongly resemble the standard MOs for an isolated water molecule.78 This close resemblance is by no means predetermined and is a validation of the EMO method. For clarity, we will continue to use the isolated atomic or molecular symmetry labels in referring to individual atomic orbitals, MOs and bands in the aqueous system.
The EMO approach is sufficiently general to be used on gas-phase systems, as well as condensed (periodic) systems, an illustrative example of the simple gas-phase (Cl− + H2O) cluster is provided in Fig S4 (ESI†). Changing the functional from BLYP to PBE does not significantly affect the results (Table S1, Fig. S5 and S6, ESI†).
BLYP is well known to be unable to match the absolute position of water orbital energy levels, and thus we apply an energy shift to align the computed water 1b2 HOMO peak maxima to that obtained experimentally, −11.16 eV.88 A comparison of orbital energies computed at the BLYP, B3LYP and MP2 levels for the simple gas-phase (Cl− + H2O) cluster is provided in Fig. S7 (ESI†). Water can be thought of as an oxide with a band gap of 8.7 eV (wide band gap insulator) which is underestimated by BLYP. Traditionally one might assume that the HOMO or valence band maximum (VBM) is correctly determined and that the error lies in the position of the LUMO or conduction band minimum (CBM). However, not only is the water CBM incorrectly referenced in BLYP but also the water VBM. Indications were already evident in 2005 that this was the case89 but this has now been firmly established.90 The origin of this error is thought to be the self-interaction or delocalisation error. It has been shown that the band gaps of crystalline insulators computed at the DFT/GGA level are substantially affected by the amount of Hartree–Fock exchange included in the exchange–correlation functional, and that not only the CBM but also the VBM are influenced by this effect.91 More recently it has been shown that the VBM of an aqueous solution is displaced more (relative to the experimental value) than the CBM.90,92 Thus the electronic position of the water HOMOs from our BLYP simulations are approximately 3.5 eV higher than they should be, and we rescale to the experimentally determined valence band, where the peak center is at −11.16 eV. More positively, the lower orbitals appear to be at roughly the correct energies when referenced to the water conduction band.88
In order to compare the magnitude of the water–chloride and water–water interaction energies, B3LYP and MP2 calculations employing a cc-pVTZ basis set have been carried out using Gaussian 09.93,94 All structures have been optimised under no symmetry constraints with enhanced convergence criteria for the energy and density, and are confirmed as minima by frequency calculations. A thermochemical analysis has been carried out to determine zero point energy (ZPE) corrections and an estimate of the basis set superposition error (BSSE) has been obtained using the counterpoise method.95
3 Results and discussion
3.1 Benchmarking: comparison of RDFs with experiment and simulation
When a Cl anion inserts into the H-bonding network of liquid water directional Cl–water H-bonds are formed. The H-bonding interactions are not static; thermal and electronic motions allow H-bonds to break and reform in new patterns. The Cl anion can “mimic” a water molecule in undertaking at least two H-bond acceptor interactions, however there is an absence of H-bond donor interactions.Radial distribution functions (RDFs) provide an average H-bond length and an indication of the variation in length for the water–water and Cl–water H-bonds. They also allow us to evaluate the quality of the simulations by comparing computed RDFs with those obtained experimentally. Relevant data from our simulation are presented in Table 1.
| r 1st maxima | r 2nd maxima | r 1st minima | CN | |
|---|---|---|---|---|
| g OO | 2.74 | 4.41 | 3.29 | 4.2 |
| g OH | Bond | 1.76 | 1.23 | — |
| g ClO | 3.15 | 4.74 | 3.83 | 6.3 |
| g ClH | 2.19 | 3.62 | 2.90 | 5.1 |
The RDFs of pure liquid water are typically well reproduced by CPMD simulations.81,83 The O–O and O–H RDFs for the chloride solution simulated here (Fig. S8 and S9, ESI†) resemble those of pure water, indicating that the Cl anion does not significantly affect the long range water structure at this concentration.83 For comparison, the experimental O–O and O–H RDF maxima for pure water are 2.88 Å and 1.85 Å respectively. The experimental first minimum in the O–O RDF occurs at 3.3 Å and the coordination number is 4.5.96
The computed Cl–O and Cl–H RDFs (Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O ratio 1:63) are presented in Fig. 1. Relevant neutron diffraction experiments have been used to study solutions bracketing our Cl:water ratio: 1:11 and 1:83 with counter ions Na+ and K+.20,21 The experimental gClO and gClH RDF maxima bracket our results. Moreover, greater variation was found when changing the counter ion, than when changing the concentration. Other experiments have found the first peak of the Cl–O RDF to be in the range of 3.01–3.36 Å varying slightly with concentration.44,56,71 The experimentally determined coordination numbers for aqueous chloride solutions vary with concentration; for ratios of 1:11–1:83 (Cl:H2O) the Cl–O and Cl–H coordination numbers vary between 5.7–7.0 and 5.9–6.1 respectively.20 The computed Cl–H coordination number is within the lower error bound (±1.1) of the experimental values. Comparison with the experimental results described above indicate that our simulation can be considered to be reflective of aqueous chloride solutions (around a concentration of 0.88 mol L−1 for K+ and Na+ monovalent counter ions).
:H2O ratio 1:63) are presented in Fig. 1. Relevant neutron diffraction experiments have been used to study solutions bracketing our Cl:water ratio: 1:11 and 1:83 with counter ions Na+ and K+.20,21 The experimental gClO and gClH RDF maxima bracket our results. Moreover, greater variation was found when changing the counter ion, than when changing the concentration. Other experiments have found the first peak of the Cl–O RDF to be in the range of 3.01–3.36 Å varying slightly with concentration.44,56,71 The experimentally determined coordination numbers for aqueous chloride solutions vary with concentration; for ratios of 1:11–1:83 (Cl:H2O) the Cl–O and Cl–H coordination numbers vary between 5.7–7.0 and 5.9–6.1 respectively.20 The computed Cl–H coordination number is within the lower error bound (±1.1) of the experimental values. Comparison with the experimental results described above indicate that our simulation can be considered to be reflective of aqueous chloride solutions (around a concentration of 0.88 mol L−1 for K+ and Na+ monovalent counter ions).
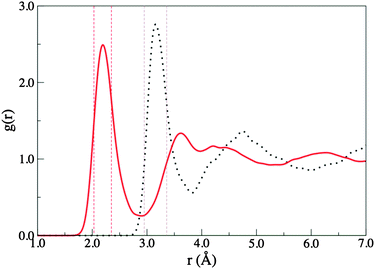 | ||
| Fig. 1 Radial distribution functions for Cl–O (dotted line) and Cl–H (solid line) for an aqueous chloride solution. Dashed lines represent the range of the first maxima of the RDF from experiments.20,44,56,71 | ||
3.2 Effective MO analysis
A unique aspect of the EMO analysis is that it allows resolution of the local orbitals associated with each molecule, solute orbitals can be resolved from the sea of solvent orbitals, and it is possible to investigate relationships between the local solvation environment and the local coordination environment.The EMOs of the Cl anion at one time step (19.32 ps) of the trajectory are plotted in Fig. 2. The effective MOs closely resemble the respective atomic and molecular orbitals of the isolated molecules with slight distortions due to the influence of neighbouring water molecules.
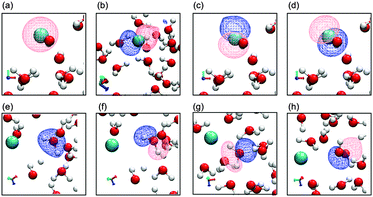 | ||
| Fig. 2 EMO plots of the Cl anion and one water molecule in the first solvation shell. Cl solute EMOs (a) 3s-like orbital and (b)–(d) 3p-like orbitals, water EMOs (e) 2a1, (f) 1b1, (g) 3a1 and (h) 1b2. | ||
In a condensed environment the individual molecular orbitals form ‘bands’. Fig. 3 shows the density of states (DOS) for liquid water with a single Cl anion impurity, computed by averaging over 80 instantaneous atomic configurations taken from an ab initio trajectory of ∼30 ps duration. The DOS of pure liquid water has been studied in detail previously using first-principles approaches and photoemission spectroscopy.78,88 The effects on the DOS of adding a Cl anion at this concentration appear to be relatively minor, and there are no large changes in position or intensity. However, the overlap of the Cl 3p and water EMOs suggests a potentially significant level of orbital mixing.
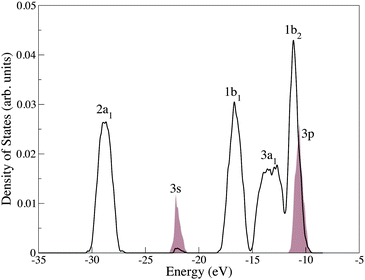 | ||
| Fig. 3 Total density of occupied states for an aqueous chloride solution. The water bands are labeled following the convention for a C2v symmetry H2O molecule. The chloride ion peaks are represented by shaded areas which, for clarity, have been magnified by a factor 10. | ||
The Cl anion can be thought of as a charged defect in a wide band gap oxide (water). The question arises of how accurately the Cl anion HOMO is positioned relative to the water VBM (given the problems BLYP has in positioning the water conduction band). Experimentally, the Cl anion detachment level is just above (−8.8 eV) the valence band edge of water (−9.9 eV) and in spectra the Cl anion 3p orbitals appear as a shoulder on the water valence band due to broadening.88,97 Solid state charge defect energies can be in error by amounts as large as 1 eV, and similar errors can occur in aqueous solution.92 This is significant as it can determine whether the Cl anion HOMO lies above or is buried within the water band, changing the character of the HOMO for the whole system. Broadening of the Cl anion HOMO (thermal and electronic) complicates the matter further, and thus the Cl anion HOMO is mixed up with the valence band of water. The extent to which the delocalisation error in BLYP affects this mixing is not known. Our results indicate that when the valence band of water is adjusted to match the experimental value, the Cl anion HOMO peak center occurs at −10.12 eV, which lies above the band maxima, but on the band edge of water. Previous calculations treating the ion at the MP2 level but the surrounding water molecules as point charges obtained 1.04 eV compared to an experimental estimate of 0.6 eV, we obtain a maximum broadening of 1.39 eV.88,97 A further application of the EMO method to a simulation in which a hybrid exchange functional has been employed, and a comparison with the results presented here would be highly insightful.
Over the course of a trajectory the electronic structure of the solute and solvent molecules varies in time. Unique to the EMO method is the ability to monitor variations of the individual effective MOs over time. Fig. 4 shows the position of the Cl anion effective HOMO relative to the water 1b2 HOMO band for selected points along a trajectory.
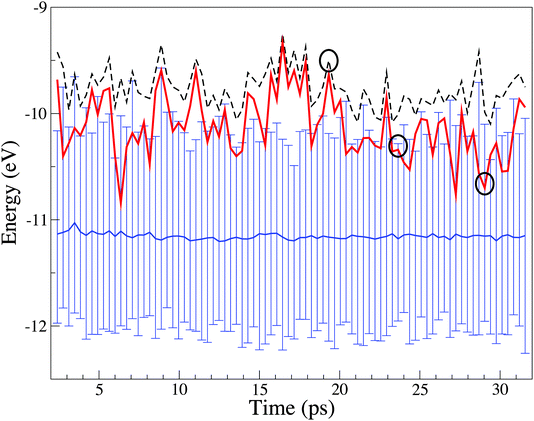 | ||
| Fig. 4 Variation in energy of the chloride HOMO and highest energy occupied water band over the course of a trajectory. The dashed line refers to the system Kohn–Sham HOMO. The thick red line refers to the effective HOMO of the Cl anion. The continuous blue line represents the centre of the valence band of water, and the vertical lines mark its instantaneous width (conventionally indicated in Fig. 3 as 1b2) representing the range of HOMO energies of the 63 water molecules in the system. The upper and lower edges of the vertical lines represent the upper and lower range of the 63 HOMO energies. The circles identified specific sampled structures. | ||
Over the course of the trajectory, three parameters α, Δα and β have been computed and averaged for the water and Cl anion EMOs, Table 2. α is the center of each effective MO band. For example, α for the 1b2 effective MO relates to the blue line at the center of the water HOMO band in Fig. 4, and it is the average of these values over the whole trajectory. Δα is the “thermal broadening” of a band due to changes in the molecular environment over time, and is related to temperature fluctuations (evident in the oscillations of the band edges). β is the electronic broadening due to overlap with orbitals from other molecules. The combined effect of the thermal and electronic broadening contribute to the total “width” of a band. Details of how these quantities are derived can be found in ref. 78.
| MO | α | Δα | β |
|---|---|---|---|
| Water | |||
| 2a1 | −28.48(−28.30) | 0.45(0.43) | 0.78(0.58) |
| 1b1 | −15.96(−15.79) | 0.52(0.46) | 1.21(1.41) |
| 3a1 | −13.11(−12.88) | 0.47(0.48) | 1.27(1.57) |
| 1b2 | −11.16(−10.90) | 0.44(0.42) | 1.01(1.11) |
| Chloride | |||
| 3s | −21.66 | 0.31 | 0.71 |
| 3p | −10.53 | 0.31 | 1.36 |
| 3p | −10.29 | 0.33 | 1.11 |
| 3p | −10.12 | 0.30 | 0.82 |
From Fig. 4 it is evident that while individual water EMOs vary significantly in energy (see the top and bottom of the water HOMO band), the average energy (band center) remains stable over the whole trajectory. The system HOMO is the “reactive” region and in the EMO analysis the HOMO can be unambiguously associated with a particular molecular species. The Cl anion dominates the system HOMO about 48% of the time, while a water molecule dominates it 52% of the time. This has implications for the instantaneous reactivity of the system. Nevertheless, this result must be treated with caution due to the propensity of BLYP to over delocalise the Kohn–Sham orbitals.
Interpreting the “energy level” or α data, the water α values in the aqueous Cl anion solution are similar to those of the pure water, and the same is true for Δα. Moreover there is not a great spread in these values. These results indicate that the Cl anion, at this concentration, has only a small effect on the water orbital energy levels or thermal broadening. This is consistent with PES experiments which show no disturbance of the electronic structure (DOS) of water for iodide salts.98
From the EMO perspective the valence band of water is composed of the 1b2 MOs of different water molecules, that is “lone pair” orbitals localised on the oxygen atoms. The experimental and the computed valence band are not substantially broadened, indicating that these orbitals remain non-bonding between water MOs. The deeper, H-bonding capable water MOs (1b1 and 3a1) show more broadening and therefore water–water interactions. The experimentally observed broadening of the deep 2a1 MO is not well recovered. Lone pairs of this type are generally considered to correlate electrons in dispersive interactions. Simulations including dispersion interactions have just recently become possible.68,99,100 A further application of the EMO method to a simulation in which a dispersion corrected functional has been employed, and a comparison with the results presented here would be useful for understanding the extent of dispersion effects on the electronic structure.
The thermal (environmental) broadening Δα for the Cl anion orbitals is ≈0.31 eV, smaller than that of the water bands. This may be due to fewer configurations sampled, 80 compared to the 5040 for the water orbitals, or a genuinely smaller Δα for the Cl anion. This indicates that the local environment for the chloride ion is more structured and that it varies less over time than for a bulk water molecule. This is consistent with the slower dynamics observed for the chloride–water compared to water–water H-bond interactions in the bulk.
The Cl anion β values are of a similar magnitude to the water molecule β values. This means that the water–water coupling is similar to the chloride–water coupling. For both the chloride ion and water molecules the HOMO shows a reduced electronic coupling compared to the slightly lower energy EMOs. This indicates that interactions are not dominated by the molecular HOMO (HOMO–LUMO interactions are often drivers for chemical reactivity). In contrast to α, the electronic broadening of the water β values is rather sensitive to the addition of a chloride ion. β also varies more strongly than Δα (standard deviations: 0.16, 0.25, 0.25 and 0.28 eV).
So far the quantities analysed have been trajectory averages, however the nature of the orbital interactions between the chloride ion and solvent water molecules can be interpreted in terms of instantaneous descriptors, α, Δα and β computed for snap-shots from the trajectory, and thus we are now in a position to link a local solvation geometry to the stability of the chloride HOMO.
Several representative structures, indicated by the black circles in Fig. 4, have been selected for a more detailed analysis, Fig. 5 and Tables S2 and S3 (ESI†). These snap-shots represent high, even and low energy chloride HOMOs relative to the water band edge. In Fig. 5, atoms within 12 Å of the chloride ion are depicted as stick structures, hydrogen atoms within the first minimum of the Cl–H RDF are represented as light grey spheres. Oxygen atoms within the first solvation shell boundary (defined by the first minimum in the Cl–O RDF) are represented as spheres, two types of water molecules are identifiable; “H-bonding” water molecules that are directly H-bonding (coordinated) to the chloride ion (red spheres) and “local” water molecules (blue spheres) that are not H-bonding to the chloride ion (hydrogen atoms outside of the first minimum of the Cl–H RDF).
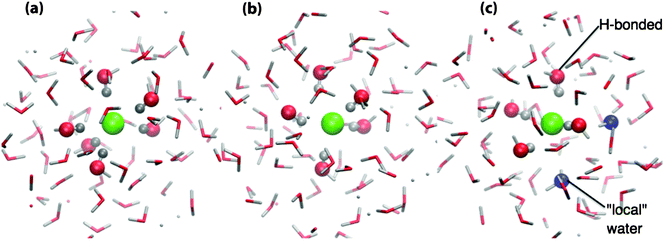 | ||
| Fig. 5 Selected configurations centered on the chloride anion and showing the first solvation shell water molecules: (a) low-energy chloride HOMO, (b) intermediate-energy chloride HOMO, and (c) high-energy chloride HOMO, see the text for a description of the atoms represented as spheres. | ||
Fig. 5(a) shows a configuration where the chloride HOMO lies deep within the water band, i.e. when Cl anion is stabilized. In this case, there are six water molecules H-bonding to the Cl anion and no local water molecules, i.e. all of the water molecules within the first solvation shell boundary are H-bonding to the Cl anion. Fig. 5(b) shows a configuration in which the top of the water HOMO band and the chloride HOMO are essentially degenerate, in this case the Cl anion is H-bonding to five water molecules arranged roughly in a distorted square-based pyramid. Fig. 5(c) shows a local configuration which produces a high energy Cl anion HOMO, in this case there are four water molecules H-bonding with the Cl anion and two local water molecules. Thus the Cl anion favours coordination by six H-bonding water molecules. The lower coordination numbers are therefore likely due to the water dynamics, such that only five or even four water molecules are available.
Using the EMO method it is possible to provide a numerical value representative of the electronic effects of the chloride ion within and beyond the first solvation shell (i.e. β). For the individual structures selected above Cl–water interactions have been divided into those with water within the first solvation shell (β1stshell–Cl), and those with water outside the first solvation shell (βbulk–Cl). Water–water interactions have been divided into bulk water interacting with either the first solvation shell (β1stshell–bulk), or bulk water (βbulk), Fig. 6 and Table 3. (A detailed breakdown of the β1stshell–Cl values for individual water molecules within the first solvation shell is given in Table S3, ESI.†)
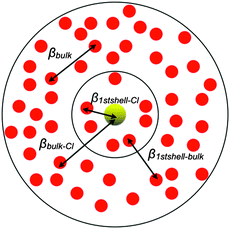 | ||
| Fig. 6 Representation of electronic interaction per pair of molecules: the light (yellow) solid circle represents the Cl anion, the inner black circle represents the first solvation shell, and the outer black circle represents bulk water. | ||
| Low Cl− HOMO 5(a) | Level Cl− water HOMOs 5(b) | High Cl− HOMO 5(c) | |
|---|---|---|---|
| β 1stshell–Cl | 0.179 | 0.206 | 0.137 |
| β bulk–Cl | 0.004 | 0.004 | 0.004 |
| β 1stshell–bulk | 0.014 | 0.014 | 0.013 |
| β bulk | 0.017 | 0.017 | 0.018 |
The data in Table 3 show that interactions between the Cl anion and water molecules of the first solvation shell, β1stshell–Cl, are much larger than the average water–water interactions, βbulk ≈ 0.017 eV. However, despite a strong interaction with the chloride ion, which might be expected to reduce the interaction with bulk water molecules, these molecules maintain a large proportion of their water–water interactions, β1stshell–bulk ≈ 0.014 eV. This is a key conclusion: water molecules which are involved in H-bonding to the chloride ion are not prevented from interacting with other water molecules.
To further investigate the decay of βCl–water away from the Cl anion, and thus to evaluate the extent of the electronic influence of the Cl anion on water molecules outside the first solvation shell, βCl–water for water molecules within 6.0 Å of the Cl anion have been evaluated for 10 snap-shots selected from the trajectory and plotted against the radial Cl–O distance, Fig. 7. The decay curve has been fitted by an exponential, β0e−λR, with β0 = 13.66 eV and λ = 1.40 Å−1; and a stretched exponential, 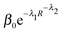 , with β0 = 57.85 eV, λ1 = 1.66 Å−1 and λ2 = 1.08. Despite the fact that a plane wave basis has been employed, the computed EMOs are localised on individual molecules. Hydrogenic orbitals decay exponentially with respect to the distance from the nucleus, and for simple hydrogenic orbital bond integrals, decay is exponential with respect to the internuclear separation. Thus obtaining an exponential decay for beta appears to be reasonable. However, the stretched exponential is clearly more indicative of the decay of the β integral with distance. We interpret this non-exponential decay as a manifestation of heterogeneous dynamics in our system, i.e. with individual relaxing units, for example, subsets of hydrogen bonded water molecules, or partially ordered solvation shells having individual relaxation times.101,102
, with β0 = 57.85 eV, λ1 = 1.66 Å−1 and λ2 = 1.08. Despite the fact that a plane wave basis has been employed, the computed EMOs are localised on individual molecules. Hydrogenic orbitals decay exponentially with respect to the distance from the nucleus, and for simple hydrogenic orbital bond integrals, decay is exponential with respect to the internuclear separation. Thus obtaining an exponential decay for beta appears to be reasonable. However, the stretched exponential is clearly more indicative of the decay of the β integral with distance. We interpret this non-exponential decay as a manifestation of heterogeneous dynamics in our system, i.e. with individual relaxing units, for example, subsets of hydrogen bonded water molecules, or partially ordered solvation shells having individual relaxation times.101,102
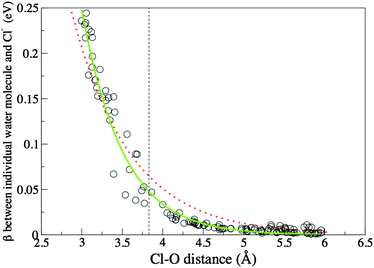 | ||
| Fig. 7 β as a function of Cl–O distance. Circles are calculated data points, the dotted (red) line is the fitted exponential, the continuous (green) line is the fitted stretched exponential, and the dashed line identifies the first minimum of the Cl–O RDF. | ||
Values of β are significantly larger than 0.004 (background bulk–Cl interaction) beyond the first solvation shell. Employing the fitted equation, β decays to the background level at 5.82 Å just before the second minimum of the Cl–O RDF (which is close to 6 Å). Thus, there is a direct electronic influence of the Cl anion into the second solvation shell, but this effect does not extend beyond the second solvation shell. The similarity of the water–water β1stshell–bulk and βbulk quantities also indicate that indirect electronic effects beyond the second solvation shell are minimal. While these results show that the effect of the Cl anion does not extend beyond the second solvation shell, these data should be treated as a maximum estimate for the water–Cl anion electronic coupling due to the BLYP functional exaggerating electronic delocalisaton.
The Cl anion can “mimic” a water molecule effectively in terms of matching the electronic overlap with other water molecules. In Fig. 7 the largest β values are for interaction of first solvation shell water molecules with the Cl anion (0.25), and analysis of the direct water–water interactions in bulk water also gives strong beta values (0.26). It might be considered that a large orbital coupling should result in some charge transfer. This would be the case for normal bonding interactions where one fragment orbital is occupied and the other unoccupied. However, in this case both fragment orbitals are occupied and significant redistribution of electron density from one molecular fragment to another is not expected.
3.3 Local solvation around the Cl anion
The coordination number and the local arrangement of the water molecules around the Cl anion is of particular interest. Based on our simulations the Cl anion is on average surrounded by 5–6 water molecules, however, the Cl–O coordination number (6.3) is essentially one more than the Cl–H derived coordination number (5.1). The larger coordination number for oxygen compared to hydrogen has also been found experimentally at the Cl:water 1:83 ratio, this was ascribed to the presence of an interstitial water molecule.20
Inspection of the trajectory shows that the hydrogen atom coordination number reflects the number of water molecules H-bonding to the Cl anion. The extra oxygen atom or atoms belong to “local” water molecules. Local water molecules have oxygen atoms that lie within the first minimum of the Cl–O RDF but hydrogen atoms that are not within the first Cl–H RDF minimum.
Typically there are 1–4 extra local water molecules, and they take on a variety of conformations. In Fig. 8 water molecules within 8.0 Å of the Cl anion are depicted, those lying within the Cl–O boundary are represented as ball and stick structures, blue oxygen atoms labelled L belong to local water molecules, purple water molecules labelled B are not within the first solvation shell but have been identified to highlight specific water molecules that are H-bonding with the local water molecules, the remaining ball and stick water molecules (with red oxygen atoms) are those that H-bond to the Cl anion, H-atoms within the Cl–H boundary are represented as dark grey.
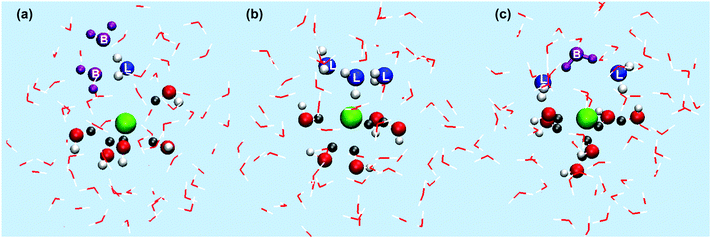 | ||
| Fig. 8 Example structures showing the local arrangement of water molecules in the first solvation shell around the Cl anion, see the main text for a full description. (a) Single local water H-bonding with bulk water, (b) multiple local water molecules mutually H-bonding, (c) two local water molecules H-bonding to coordinated water molecules. | ||
For example, a local water molecule can H-bond to the surrounding bulk water, rotated such that the H-atoms are facing away from the chloride ion and interacting with bulk water, Fig. 8(a). Fig. 8(b) shows multiple local water molecules H-bonding to each other, and Fig. 8(c) shows local water molecules H-bonding to water molecules H-bonding with the Cl anion. Visual inspection of the trajectory shows that first solvation shell water molecules predominantly collect on one side of the Cl anion, often but not always, the local water molecules collect on the opposite side, H-bonding to each other or bulk water. The Cl anion H-bonding water molecules tend not to H-bond with each other, but form H-bonds with other bulk water molecules.
The number of water molecules associated with the chloride ion has been evaluated over time. Fig. 9 shows a histogram of the coordination numbers collected over the trajectory based on the average Cl–O and Cl–H distances defined in Table 1. The H-atom distribution is narrower and dominated by a coordination number of five. From the Cl–O distribution it is evident that the Cl anion is associated ≈25% of the time with 5 water molecules and ≈40% of the time with 6 water molecules; this means that there is regularly one local water molecule present in the first solvation shell. However, larger numbers of local water molecules can occur; ≈30% of the time there are 7–9 water molecules within the first solvation shell.
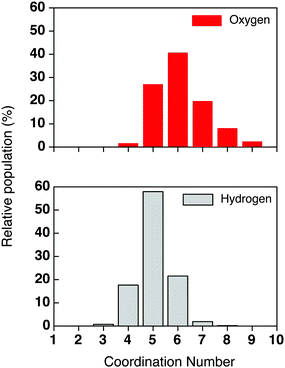 | ||
| Fig. 9 Histogram of the coordination number based on the Cl–O and Cl–H distances for Cl− in water solution. | ||
The distribution of O–Cl–O angles in the first solvation shell is shown in Fig. 10. Fig. 10 shows a maximum around 80°, and small peak around 46°, consistent with water molecules in the first solvation shell collecting on one side of the chloride ion. A similar asymmetric distribution has been found for ab initio MD simulations of aqueous bromide or chloride ions,67,103 and neutron diffraction studies have found a peak at about 70°.20 It is also well known that in small clusters (or at an interface) the Cl ion prefers an unsymmetrical solvation shell.65
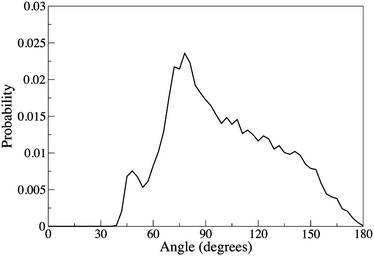 | ||
| Fig. 10 Distribution of the O–Cl–O angle calculated within the first minimum of the Cl–O RDF. | ||
To investigate differences with respect to gas phase clusters, a small sample of representative calculations was carried out at the B3LYP/cc-pVTZ and MP2/cc-pVTZ levels for selected conformers of Cl(H2O)5− and Cl(H2O)6− (Tables S5 and S6 and Fig. S10–S12, ESI†). [Cl(H2O)5]− is slightly more stable than [Cl(H2O)4·H2O]− by ≈5 kJ mol−1. [Cl(H2O)6]− with all the water molecules H-bonding in a ring below the chloride ion was unstable (Fig. S12a, ESI†) and a distorted octahedral geometry was preferred (Fig. S11a, ESI†). This is consistent with the lowest energy Cl anion solvation shell structure extracted from the trajectory. Multiple structures with [Cl(H2O)4·(H2O)2]− and [Cl(H2O)3·(H2O)3]− arrangements were found, some close in energy to the most stable structure (between 0 and 20 kJ mol−1). Thus, structures with bridging and second solvation shell water molecules are also evident in gas-phase clusters. The stability of an additional Cl–water H-bond is similar to that obtained from a water–water H-bond. A key difference is that the first solvation shell water molecules in the gas phase clusters are forced to form H-bonds with each other, while the liquid phase first solvation shell water molecules prefer to H-bond with the second solvation shell or bulk water molecules. More importantly “local” water molecules not H-bonding to the Cl anion cannot occur for the clusters, as they need to be stabilised by H-bonding to second solvation shell water molecules.
The HOMO, LUMO energy levels and HOMO–LUMO gap are presented in Tables S7 and S8 (ESI†). The energy levels from the MP2 calculations are closer to those obtained experimentally for the liquid, however B3LYP provides a better estimate of the HOMO–LUMO gap. The position of the Cl anion HOMO relative to the water valence band edge (VBE) in the B3LYP and MP2 calculations for these selected conformers has also been computed (Tables S7 and S8, ESI†). In all cases, the HOMO of the clusters is of 3-fold degeneracy on the Cl anion, and the VBE of water lies below the Cl anion HOMO. For example, for the most stable conformer of Cl(H2O)6−, MP2 calculations predict that the HOMO (−7.04 eV) of the Cl anion is 2.71 eV above the VBE of water (−9.75 eV); B3LYP calculations predict the HOMO of the Cl anion and the water VBE to be −3.40 eV and −4.75 eV, respectively; producing an energy gap of 1.35 eV. Thus, the B3LYP prediction is compared more favourably to the corresponding experimental energy gap (1.1 eV)97 within the liquid.
3.4 Dipole moments
The Cl anion is polarized within the bulk liquid; directional H-bonds are formed, and water molecules “group” on one side of the Cl anion. These interactions are not static, H-bonds are breaking and reforming in new patterns, and the polarisation of the Cl anion shifts. Not only does the water respond to the anion, but the anion responds to the water. Thus, a robust description of the dipole and electronic polarizability is important for studying ionic systems, and this is naturally taken into account by the electronic wavefunction of the ab initio MD.Dipole moments have been computed employing a Wannier function analysis. The Wannier orbitals are obtained from the Kohn–Sham orbitals via a unitary transformation, where the spread of the orbitals is minimized iteratively.104 Using the centres of the Wannier functions and associated charges, the dipole moment of a water molecule can be calculated using the classical dipole moment formula. This type of analysis has previously been employed to analyse dipole moments.70,103,105–110
The calculated dipole moments of the water molecules inside and outside the first solvation shell are 3.05 ± 0.14 and 3.10 ± 0.06 D, respectively, and thus they are essentially equal within the error limits. Our results are consistent with those from previous CPMD studies67,68,81,103,111 and an experimentally based estimate for the dipole moment of pure water, 2.9 ± 0.6 D.112 The halide and concentration of the other simulations does vary, however even in concentrated solutions the average dipole moment of water does not appear to vary by a large amount, for example an average dipole moment of 3.07 D has been obtained for water in a 14 mol L−1 solution of LiCl.113 The calculated dipole moment of the Cl ion is 0.79 ± 0.39 D. This value compares favourably with the results from other CPMD simulations 0.82 ± 0.32 D,68 and the value from a combined quantum chemical and statistical model of 0.35 ± 0.14 D seems to be low.114
Heuft and Meijer found significant fluctuations in the chloride dipole moment, however only 13 configurations were sampled, in most of these the dipole was found to be ≈1 D but there were two configurations with much larger dipole moments, up to 4 and 5 D.67 We have sampled 80 configurations, and the chloride ion dipole moment deviates significantly from zero over the simulation, Fig. 11. However, we found no dipole moments of magnitude greater than 1.75 D. A similar study for the bromide ion found a dipole moment of 0.95 with a standard deviation of 0.8 D, and a maximum dipole of ≈1.75 D.103
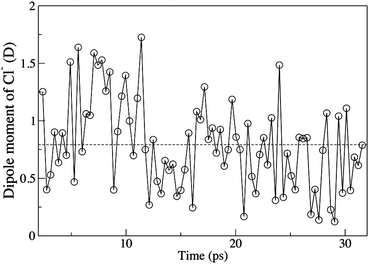 | ||
| Fig. 11 Time evolution of the dipole moment of the aqueous chloride ion, the dashed line is the average dipole moment. | ||
Importantly, the average magnitude of the chloride ion dipole is ≈3 times less than that of the water molecule it “replaces”. The chloride ion is less “polarized” than a neutral water molecule. This is consistent with water molecules in small chloride ion water clusters having a smaller dipole moment; the chloride ion is less polarizing than another water molecule. For example, the dipole moment of water in small clusters Cl(H2O)n (n = 1–5) has been found to be in the region of 2.5–2.6 D.70 This difference has been taken to mean that dipole moments for water molecules in the first solvation shell are dominated by water–water rather than ion–water interactions.
In the past, the ability of the chloride ion to form “a large dipole” has been suggested as the driving force for the formation of stable cluster geometries with the water molecules collected on one side of the ion.63 However, the small dipole moment determined for the chloride ion relative to the water molecules does not align well with this rationalisation. The polarizability of the chloride ion relative to water molecules must play a significant role. The small dipole moment induced on the chloride ion may not be the only driver for an asymmetric solvation shell. A highly symmetric, non-polarised cluster, or first solvation shell is not favoured entropically, and a larger range of configurations and H-bonding motifs can occur if the chloride ion takes a polarised configuration. The stabilisation of water–water H-bonds is important, and the small number of water molecules in a cluster means that they must collect on one side of the chloride in order to form water–water H-bonds. This limitation is removed in the aqueous system where H-bonding with bulk (second solvation shell) water molecules is possible, and as shown above favoured.
As the dipole moment of the whole system is zero, fluctuations of the chloride ion dipole must be accommodated by the water environment. Because the dipole moments of first solvation shell water molecules do not differ significantly from those in the bulk water, these adjustments must be small. The results presented here show that the average water dipole moment is not strongly affected by the presence of the chloride ion. Fig. S13 (ESI†) shows how a water molecule can maintain a directional H-bond to the chloride ion while still allowing the water molecule to re-orientate around the H-bonded H–O bond vector, leaving the molecular dipole moment free to re-orientate. Such effects result in different dynamics for H-bonds and dipole moments.115
3.5 Exchange events
A residence time reflects the time a water molecule remains H-bonded to the chloride ion. Residence times computed for water near a chloride ion can vary depending on the method employed, for example a time constant of 4.5 ps has been obtained from classical MD (employing a non-polarizable potential),56 while including polarization effects increases this to 2–3 times longer,24 and employing ab initio MD a residence time of 12 ± 3 ps has been obtained.67 The spectral diffusion time for the OH stretch (i.e. fluctuations in the length of the O–H bond) are thought to be related to the residence time of the water molecule. Using femtosecond pump–probe IR spectroscopy the time constant for bulk water is 0.5 ± 0.2 ps and for water in the first solvation shell of a chloride anion was determined as 12 ± 3 ps.52 Using self-diffusion constants determined from NMR the residence time for a water molecule in the hydration shell of chloride is 15 ps.24 This match between computed and experimental time constants indicates that the ab initio MD is recovering the dynamics of water molecules around a chloride anion.Events such as water molecules leaving or entering the first solvation shell are rare, but can be monitored by examining the Cl–O radial distances of the water molecules (Fig. 12 shows a single event, Fig. S14 (ESI†) shows the Cl–O radial distance for all the water molecules along the course of the trajectory). Two time scales are evident: rapid oscillations which reflect H-bonds breaking and reforming, and slower dynamics which show when molecules cross the first or second solvation shell boundaries. During our simulation 10 water molecules moved between the first and the second solvation shells, and both timescales are readily observable in the animated trajectory. There are experimental decay constants supporting these observations; water molecules break and reform H-bonds (2.6 ps), and reorientate (9.6 ps) much faster than they dissociate from the anion (≈12 ps).51,52
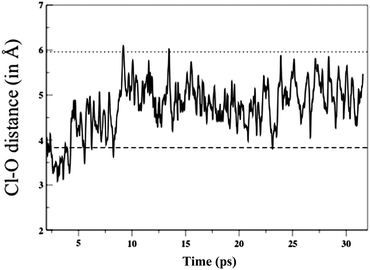 | ||
| Fig. 12 Time evolution of the Cl–O distance for a single water molecule, the dashed lines are the first and second solvation shell boundaries. | ||
The movements of the ten water molecules exchanging around the chloride ion can be divided into three categories, representative examples of the evolution of the radial distances are shown in Fig. S15 (ESI†). (i) A water molecule stays in the first solvation shell for ≈6 ps, then leaves, remains in the second solvation shell or comes back to the first solvation shell, there are three molecules in this category. (ii) A water molecule enters the first solvation shell briefly and then leaves, there are five molecules in this category. (iii) A water molecule enters the first solvation shell and remains in the shell for the rest of the simulation, there are two molecules in this category. In some of these cases a water molecule has entered the first solvation shell and a short time later a different water molecule leaves. Overall one water molecule remained in the first solvation shell over the entire trajectory, four water molecules moved into the first solvation shell and stayed, three water molecules left and returned, and two water molecules moved out of the first solvation shell not to return.
Inspection of the trajectory shows that water molecules with a H-bond to the Cl anion tend to remain close to the Cl anion. The local water molecules (those within the first Cl–O boundary but not H-bonding to the Cl anion) are more dynamic. In analogy with the phenomenon of “slaving” observed in protein dynamics,101,102 these water molecules may respond more readily to bulk water fluctuations. Hence, an analysis based purely on the Cl–O radial distance of water molecules within the first solvation shell may not provide sufficient resolution, there may be two time-scales for water molecules around the Cl anion, one for directly H-bonding and one for local water molecules. In this case a dual selection criteria including Cl–H and Cl–O radial cut-off may offer more insight. The EMO β analysis indicates that while the local water molecules (β ≈ 0.05) are not as strongly interacting as the H-bound water (β ≈ 0.20) they are interacting with the chloride ion more than bulk water molecules (β ≈ 0.004).
Inspection of the motions occurring around the Cl anion (Fig. S16, ESI†) shows small changes in distances and angles. Motion of the H-bound water molecules is more localised, and a “circling” pattern is evident. Motion of the water molecules between the Cl–O boundary and 5.0 Å from the Cl anion is less constrained and “linear” translation features are more pronounced. Motion of the local water molecules lies between these two extremes: circling or linear translation. Local water molecules tend to cross and recross the Cl–O boundary relatively freely, consistent with the breaking and reforming of water–water H-bonds. However, they also tend to stay within 5.0 Å of the Cl anion.
The formation and breaking of H-bonds and reorientation (angular changes) of water molecules are known to be related in a complex way. Unraveling the information obtained from simulation and experimental studies therefore becomes difficult. The mechanism for breaking and reforming of H-bonds in aqueous solution has not been established. In a “diffusive” mechanism, the O⋯H–O H-bond can break and then the dangling H–O subsequently undertakes small uncorrelated/random angular steps, finding some time later another partner to H-bond with. In the “angle hopping” mechanism, two water molecules become equidistant from the solute water molecule, one H-bond breaks and almost instantaneously another H-bond forms via a large angle jump, the mechanism is concerted.115,116 In addition, a large angle rotation can take a water molecule from the first solvation shell into the second solvation shell changing the coordination number of the anion.117 The motions we observe in the water molecules around the chloride ion are not easily separable into translational vs. angular components and cannot readily be used to obtain information on either of these models for H-bonding.
Thus these findings support a picture of highly mobile water molecules in the Cl anion first solvation shell. Water molecules are either relatively strongly bound to the anion by hydrogen bonds, or are only weakly kept in place by dipole interactions or by hydrogen bonds with other water molecules. The instantaneous dynamics of the local water molecules can therefore be strongly coupled with the fluctuation of molecules belonging to the second and subsequent solvation shells.
4 Conclusions
We have performed ab initio MD simulations and a geometric and orbital analysis of a Cl anion solution equivalent to a concentration of 0.88 mol L−1. The EMO analysis has allowed us to gain valuable insight into the electronic structure of a solvated ion immersed in solute orbitals. Using the BLYP functional we observe an oscillating evolution of the chloride ion HOMO at the band edge of the highest occupied water band. The robustness of this result is hard to evaluate because of the self-interaction error of BLYP, which may over delocalise the chloride states and position their energies incorrectly relative to the water VBM. However, as also evident from the experimental PES the Cl anion HOMO band is broadened and overlaps the water band edge. The difference in energy is of the order of 1 eV, which is also the order of the anticipated error from employing the BLYP functional. These results provide a good indication for the maximum extent of electronic mixing, the EMO method is general and can, in the future, be applied to functionals that have the self-interaction and long range dispersion corrected.Comparison with pure water showed that the Cl anion has little effect on the average water electronic structure. Moreover, the Cl anion has a similar orbital energy distribution and electronic coupling compared to the water molecules. In terms of electronic structure the Cl anion is “water-like”. However, the average magnitude of the Cl anion dipole is approximately three times less than that of the water molecule it “replaces”. The EMO and cluster analysis showed that the Cl anion prefers a symmetric six coordinate environment with a low dipole moment. However, the coordinating water molecules would probably prefer the Cl anion to mimic a water molecule better, and have a higher dipole moment. We found that the potential for multiple structural variations (increased entropy) and the facilitation of water–water H-bonds also favour asymmetric solvation. While water molecule H-bond dynamics are reduced when H-bonding to a Cl anion, there appears to be little restriction on the dipole moment of water molecules within the first solvation shell, and the computed dipole moments for water molecules inside or outside of the first solvation shell are the same.
While the ionic nature of the Cl anion does not induce a strong response in the solvating water dipoles, the Cl anion does have an (electronic) effect beyond the first solvation shell via Cl–water electronic coupling. β1stshell–Cl interactions are much larger than the average βCl–bulk or water–water interactions. Nevertheless, β values decay to background levels within the second solvation shell. This result adds further information to the debate on the long range effects of fully solvated ions. H-bonding with the Cl anion does not interfere with the water–water H-bonding, and β1stshell–bulk and βbulk are similar.
The local solvation structure was examined in more detail using traditional descriptors. Our initial investigation was driven by an observed difference in the Cl–O and Cl–H coordination numbers (6.3 and 5.1 respectively). This led to a differentiation between two types of water molecule within the first solvation shell. There are on average 5 water molecules H-bonding with the Cl anion, and in addition, 1–4 local water molecules which have oxygen atoms within the first minimum of the Cl–O RDF but hydrogen atoms that are not within the first minimum of the Cl–H RDF. The H-bonding water molecules tend to be distributed on one side of the Cl anion, however they do not H-bond with each other but with water molecules in the second solvation shell. The local water molecules exhibit a variety of structural motifs, in addition their position is not as localised as that of the coordinating water molecules. They can bridge coordinated water molecules, H-bond with second solvation shell water molecules or they can form a mutually H-bonded group. There is a reasonable chance that the local water molecules will be on the opposite side of the Cl anion from the coordinated water molecules.
The Cl anion appears to be more stable when symmetrically solvated by 6 H-bonding water molecules (both as a cluster and in solution), and the solution HOMO is driven up as this number decreases. The EMO analysis, similarity of the water and Cl anion electronic structure, cluster calculations, and spread of coordination numbers around 4–7 indicate that there is a fine balance between Cl–water and water–water H-bonding and orbital interactions. Thus, we can rationalise the flexibility of the chloride solvation shell structure (particularly the wide range of coordination numbers obtained experimentally) and the placement of the chloride ion near the centre of the Hofmeister series.
We also observed, in contrast to the clusters, that water molecules in the first solvation shell tend not to H-bond with each other but to bulk water molecules. These results indicate that the Cl anion solvation structure is highly influenced by the dynamics of the surrounding water, which appears to favour five asymmetrically distributed H-bonding water molecules within the first solvation shell (the Cl–H coordination number is five ≈60% of the time) and a number of local water molecules. Within the first solvation shell the solvating and local water molecules have different dynamics. Visual inspection of the trajectory and monitoring of the evolution of the radial Cl–O distance for all water molecules showed that the H-bonding water associated with the chloride ion for longer periods, while the local water molecules are more mobile. Cl anion H-bonding water molecules have a slower, more circular motion, while local water molecules had a propensity for a more linear motion. Ten molecules left or entered the first solvation shell, however rapid oscillations and the complexity of the coupled motions inhibited the identification of a clear “reaction” coordinate.
We have demonstrated the potential of the EMO method as an interpretative tool to investigate solvation at a fundamental, quantum mechanical level. We have characterised the structural and electronic factors determining the effect of a chloride ion on the presence of the surrounding polarizable water solvent, and vice versa the effect of the water on the chloride ion.
Acknowledgements
L.G. and P.H. acknowledge support from the Leverhulme Trust (F/07 058/BB). P.H. acknowledges The Royal Society for a RSURF. We sincerely thank Prof. Michiel Sprik for his contributions and stimulating discussions.References
- E. M. Knipping, M. J. Lakin, K. L. Foster, P. Jungwirth, D. J. Tobias, R. B. Gerber, D. Dabdub and B. J. Finlayson-Pitts, Science, 2000, 288, 301 CrossRef CAS.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353 CrossRef CAS.
- K. L. Foster, R. A. Plastridge, J. W. Bottenheim, P. B. Shepson, B. J. Finlayson-Pitts and C. W. Spicer, Science, 2001, 291, 471 CrossRef CAS.
- B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2009, 11, 7760 RSC.
- J. Walker, J. Wills and J. Reid, Weather, 2011, 66, 165 CrossRef.
- S. Solomon, M. M. D. Qin, Z. Chen, M. Marquis, K. B. Averyt, M. Tignor and H. L. Miller, Climate Change 2007: The Physical Science Basis, Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change (IPCC), Cambridge University Press, Cambridge, UK and New York, NY, 2007.
- NASA, http://science.nasa.gov/earth-science/oceanography/physical-ocean/salinity/, 2011.
- H. E. Beekman, H. G. M. Eggenkamp and C. A. J. Appelo, Appl. Geochem., 2011, 26, 257 CrossRef CAS.
- D. F. Parsons, M. Bostrom, P. L. Nostro and B. W. Ninham, Phys. Chem. Chem. Phys., 2011, 13, 12352–12367 RSC.
- K. J. Tielrooij, S. T. van der Post, J. Hunger, M. Bonn and H. J. Bakker, J. Phys. Chem. B, 2011, 115, 12638–12647 CrossRef CAS.
- F. Hofmeister, Naunyn-Schmiedeberg's Arch. Pharmacol., 1888, 24, 247–260 Search PubMed.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505 CrossRef CAS.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS.
- Y. Marcus, Pure Appl. Chem., 2010, 82, 1889–1899 CrossRef CAS.
- Y.-S. Lin, B. M. Auer and J. L. Skinner, J. Chem. Phys., 2009, 131, 144511 CrossRef.
- D. J. Tobias and J. C. Hemminger, Science, 2008, 319, 1197–1198 CrossRef CAS.
- P. Jungwirth and D. Tobias, Chem. Rev., 2006, 106, 1259–1281 CrossRef CAS.
- M. D. Baer, I.-F. W. Kuo, H. Bluhm and S. Ghosal, J. Phys. Chem. B, 2009, 113, 15843–15850 CrossRef CAS.
- D. Paschek and R. Ludwig, Angew. Chem., Int. Ed., 2011, 50, 352–353 CrossRef CAS.
- R. Mancinelli, A. Botti, F. Bruni, M. A. Ricci and A. K. Soper, J. Phys. Chem. B, 2007, 111, 13570–13577 CrossRef CAS.
- R. Mancinelli, A. Botti, F. Bruni, M. A. Ricci and A. K. Soper, Phys. Chem. Chem. Phys., 2007, 9, 2959–2967 RSC.
- A. Botti, F. Bruni, S. Imberti, M. Ricci and A. Soper, J. Chem. Phys., 2004, 120, 10154–10162 CrossRef CAS.
- A. Botti, F. Bruni, S. Imberti, M. Ricci and A. Soper, J. Chem. Phys., 2004, 121, 7840–7848 CrossRef CAS.
- H. J. Bakker, Chem. Rev., 2008, 108, 1456–1473 CrossRef CAS.
- A. Omta, M. Kropman, S. Woutersen and H. Bakker, J. Chem. Phys., 2003, 119, 12457–12461 CrossRef CAS.
- A. Omta, M. Kropman, S. Woutersen and H. Bakker, Science, 2003, 301, 347–349 CrossRef CAS.
- K. J. Tielrooij, N. Garcia-Araez, M. Bonn and H. J. Bakker, Science, 2010, 328, 1006–1009 CrossRef CAS.
- K. D. Collins, G. W. Neilson and J. E. Enderby, Biophys. Chem., 2007, 128, 95–104 CrossRef CAS.
- C. Cappa, J. Smith, B. Messer, R. Cohen and R. Saykally, J. Phys. Chem. B, 2006, 110, 5301–5309 CrossRef CAS.
- J. D. Smith, R. J. Saykally and P. L. Geissler, J. Am. Chem. Soc., 2007, 129, 13847–13856 CrossRef CAS.
- S. Park and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16731–16738 CrossRef CAS.
- D. A. Turton, J. Hunger, G. Hefter, R. Buchner and K. Wynne, J. Chem. Phys., 2008, 128, 161102 CrossRef.
- D. E. Moilanen, D. Wong, D. E. Rosenfeld, E. E. Fenn and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 375–380 CrossRef CAS.
- W. Wachter, S. Fernandez, R. Buchner and G. Hefter, J. Phys. Chem. B, 2007, 111, 9010–9017 CrossRef CAS.
- A. K. Soper and K. Weckstrom, Biophys. Chem., 2006, 124, 180–191 CrossRef CAS.
- J. Batchelor, A. Olteanu, A. Tripathy and G. Pielak, J. Am. Chem. Soc., 2004, 126, 1958–1961 CrossRef CAS.
- Y. Marcus, Chem. Rev., 2009, 109, 1346–1370 CrossRef CAS.
- D. Horinek, A. Herz, L. Vrbka, F. Sedlmeier, S. I. Mamatkulov and R. R. Netz, Chem. Phys. Lett., 2009, 479, 173–183 CrossRef CAS.
- B. L. Eggimann and J. I. Siepmann, J. Phys. Chem. C, 2008, 112, 210–218 CAS.
- C. D. Wick, J. Chem. Phys., 2008, 131, 084715 CrossRef.
- D. E. Otten, P. R. Shaffer, P. L. Geissler and R. J. Saykally, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 701–705 CrossRef CAS.
- D. H. Powell, A. C. Barnes, J. E. Enderby, G. W. Neilson and P. S. Salmon, Faraday Discuss. Chem. Soc., 1988, 85, 137 RSC.
- H. Ohtaki and T. Radnai, Chem. Rev., 1993, 93, 1157 CrossRef CAS.
- A. Tongraar, J. T-Thienprasert, S. Rujirawat and S. Limpijumnong, Phys. Chem. Chem. Phys., 2010, 12, 10876 RSC.
- M. Kiriukhin and K. Collins, Biophys. Chem., 2002, 99, 155–168 CrossRef CAS.
- P. N. Perera, B. Browder and D. Ben-Amotz, J. Phys. Chem. B, 2009, 113, 1805–1809 CrossRef CAS.
- M. Arshadi, R. Yamdagni and P. Kebarle, J. Phys. Chem., 1970, 74, 1475 CrossRef CAS.
- K. Hiraoka, S. Mizuse and S. Yamabe, J. Phys. Chem., 1988, 92, 3943 CrossRef CAS.
- G. Markovich, S. Pollack, R. Giniger and O. Cheshnovsky, J. Chem. Phys., 1994, 101, 9344 CrossRef CAS.
- M. F. Kropman and H. J. Bakker, Science, 2001, 291, 2118 CrossRef CAS.
- M. Kropman, H. Nienhuys and H. Bakker, Phys. Rev. Lett., 2002, 88, 077601 CrossRef CAS.
- H. Bakker, M. Kropman and A. Omta, J. Phys.: Condens. Matter, 2005, 17, S3215–S3224 CrossRef CAS.
- D. A. Turton, J. Hunger, G. Hefter, R. Buchner and K. Wynne, J. Chem. Phys., 2008, 128, 161102 CrossRef.
- P. Åke Bergström, J. Lindgren and O. Kristiansson, J. Phys. Chem., 1991, 95, 8575 CrossRef.
- M. Patra and M. Karttunen, J. Comput. Chem., 2004, 25, 678–689 CrossRef CAS.
- R. W. Impey, P. A. Madden and I. R. McDonald, J. Phys. Chem., 1983, 87, 5071 CrossRef CAS.
- A. Chandra, Phys. Rev. Lett., 2000, 85, 768 CrossRef CAS.
- L. X. Dang, J. E. Rice, J. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1991, 113, 2481 CrossRef CAS.
- W. L. Jorgensen and D. L. Severance, J. Chem. Phys., 1993, 99, 4233 CrossRef CAS.
- D. E. Smith and L. X. Dang, J. Chem. Phys., 1994, 100, 3757 CrossRef CAS.
- S. J. Stuart and B. J. Berne, J. Phys. Chem., 1996, 100, 11934 CrossRef CAS.
- I.-C. Yeh, L. Perera and M. L. Berkowitz, Chem. Phys. Lett., 1997, 264, 31 CrossRef CAS.
- L. X. Dang and D. E. Smith, J. Chem. Phys., 1993, 99, 6950 CrossRef CAS.
- L. Perera and M. L. Berkowitz, J. Chem. Phys., 1996, 96, 8288 CrossRef.
- D. J. Tobias, P. Jungwirth and M. Parrinello, J. Chem. Phys., 2001, 114, 7036 CrossRef CAS.
- J. L. Fulton, G. K. Schenter, M. D. Baer, C. J. Mundy, L. X. Dang and M. Balasubramanian, J. Phys. Chem. B, 2010, 114, 12926–12937 CrossRef CAS.
- J. M. Heuft and E. J. Meijer, J. Chem. Phys., 2003, 119, 11788 CrossRef CAS.
- E. Guardia, I. Skarmoutsos and M. Masia, J. Chem. Theory Comput., 2009, 5, 1449 CrossRef CAS.
- B. S. Mallik and A. Chandra, J. Chem. Phys., 2011, 387, 48–55 CAS.
- C. Krekeler, B. Hess and L. D. Site, J. Chem. Phys., 2006, 125, 054305 CrossRef.
- A. Tongraar and B. M. Rode, Phys. Chem. Chem. Phys., 2003, 5, 357 RSC.
- J. Chandrasekhar, D. C. Spellmayer and W. L. Jorgensen, J. Am. Chem. Soc., 1984, 106, 903 CrossRef CAS.
- E. Clementi, R. Barsotti, J. Fromm and R. O. Watts, Theor. Chim. Acta, 1976, 43, 101 CrossRef CAS.
- K. Heinzinger, Pure Appl. Chem., 1985, 57, 1031 CrossRef CAS.
- J. A. N. F. G. A. Ignaczak and M. N. D. S. Cordeiro, Electrochim. Acta, 1999, 45, 659 CrossRef.
- S. Koneshan, J. C. Rasaiah, R. M. Lynden-Bell and S. H. Lee, J. Phys. Chem. B, 1998, 102, 4193–4204 CrossRef CAS.
- M. Mezei and D. L. Beveridge, J. Phys. Chem., 1981, 74, 6902 CrossRef CAS.
- P. Hunt, M. Sprik and R. Vuilleumier, Chem. Phys. Lett., 2003, 376, 68 CrossRef CAS.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS.
- J. Hutter, et al., CPMD 3.13.2, IBM Research Division and MPI Festkoerperforschung Stuttgart, 2008 Search PubMed.
- P. L. Silvestrelli and M. Parrinello, J. Chem. Phys., 1999, 111, 3572 CrossRef CAS.
- M. Sprik, J. Hutter and M. Parrinello, J. Chem. Phys., 1996, 105(3), 1142 CrossRef CAS.
- I.-F. W. Kuo, C. J. Mundy, M. J. McGrath, J. I. Siepmann, J. VandeVondele, M. Sprik, J. Hutter, B. Chen, M. L. Klein, F. Mohamed, M. Krack and M. Parrinello, J. Phys. Chem. B, 2004, 108, 12990 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef.
- B. Winter and M. Faubel, Chem. Rev., 2006, 106, 1176 CrossRef CAS.
- P. Hunt and M. Sprik, ChemPhysChem, 2005, 6, 1805–1808 CrossRef CAS.
- J. Cheng and M. Sprik, Phys. Chem. Chem. Phys., 2012, 14, 11245–11267 RSC.
- A. Alkauskas, P. Broqvist and A. Pasquarello, Phys. Status Solidi B, 2011, 248, 775–789 CrossRef CAS.
- C. Adriaanse, J. Cheng, V. Chau, M. Sulpizi, J. VandeVondele and M. Sprik, J. Phys. Chem. Lett., 2012, 3, 3411–3415 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc, Wallingford CT, 2009 Search PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100(4), 2975 CrossRef CAS.
- H.-C. Chang, J.-C. Jiang, J.-C. Su, C.-Y. Chang and S. H. Lin, J. Phys. Chem. A, 2007, 111, 9201–9206 CrossRef CAS.
- A. Soper and M. Phillips, Chem. Phys., 1986, 107, 47–60 CrossRef CAS.
- B. Winter, R. Weber, I. V. Hertel, M. Faubel, P. Jungwirth, E. C. Brown and S. E. Bradforth, J. Am. Chem. Soc., 2005, 127, 7203–7214 CrossRef CAS.
- R. Seidel, S. Thuìřmer and B. Winter, J. Phys. Chem. Lett., 2011, 2, 633–641 CrossRef CAS.
- I.-C. Lin, A. P. Seitsonen, I. Tavernelli and U. Rothlisberger, J. Chem. Theory Comput., 2012, 8, 3902–3910 CrossRef CAS.
- Z. Ma, Y. Zhang and M. E. Tuckerman, J. Chem. Phys., 2012, 137, 044506 CrossRef.
- R. Richert, J. Phys.: Condens. Matter, 2002, 14, R703 CrossRef CAS.
- H. Frauenfelder, G. Chen, J. Berendzen, P. W. Fenimore, H. Jansson, B. H. McMahon, I. R. Stroe, J. Swenson and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5129 CrossRef CAS.
- S. Raugei and M. Klein, J. Chem. Phys., 2002, 116, 196–202 CrossRef CAS.
- P. L. Silvestrelli, N. Marzari, D. Vanderbilt and M. Parrinello, Solid State Commun., 1998, 107, 7 CrossRef CAS.
- N. Marzari and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 12847 CrossRef CAS.
- D. Bucher and S. Kuyucak, J. Phys. Chem. B, 2008, 112, 10786 CrossRef CAS.
- D. Bucher and S. Kuyucak, Chem. Phys. Lett., 2009, 477, 207 CrossRef CAS.
- J. K. Gregory, D. C. Clary, K. Liu, M. G. Brown and R. J. Saykally, Science, 1997, 275, 814 CrossRef CAS.
- L. Bernasconi, J. Blumberger, M. Sprik and R. Vuilleumier, J. Chem. Phys., 2004, 121, 11885 CrossRef CAS.
- C. Terrier, P. Vitorge, M.-P. Gaigeot, R. Spezia and R. Vuilleumier, J. Chem. Phys., 2010, 133, 044509 CrossRef.
- K. Laasonen, M. Sprik, M. Parrinello and R. Car, J. Chem. Phys., 1993, 99, 9080–9089 CrossRef CAS.
- Y. S. Badyal, M.-L. Saboungi, D. L. Price, S. D. Shastri, D. R. Haeffner and A. K. Soper, J. Chem. Phys., 2000, 112, 9206–9208 CrossRef CAS.
- L. Petit, R. Vuilleumier, P. Maldivi and C. Adamo, J. Chem. Theory Comput., 2008, 4, 1040–1048 CrossRef CAS.
- A. Öhrn and G. Karlström, J. Phys. Chem. B, 2004, 108, 8452–8459 CrossRef.
- D. Laage and J. T. Hynes, J. Phys. Chem. B, 2008, 112, 14230–14242 CrossRef CAS.
- D. Laage and J. T. Hynes, Science, 2006, 311, 832–835 CrossRef CAS.
- D. Laage and J. T. Hynes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11167–11172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp50652e |
| This journal is © the Owner Societies 2013 |