 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of spectator species on the interaction of H2O2 with platinum – implications for the oxygen reduction reaction pathways†
Ioannis
Katsounaros
*a,
Wolfgang B.
Schneider
bc,
Josef C.
Meier
a,
Udo
Benedikt
b,
P. Ulrich
Biedermann
a,
Angel
Cuesta
d,
Alexander A.
Auer
b and
Karl J. J.
Mayrhofer
*a
aDepartment of Interface Chemistry and Surface Engineering, Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Strasse 1, D-40237 Düsseldorf, Germany. E-mail: katsounaros@mpie.de; mayrhofer@mpie.de; Fax: +49 211 6792 218; Tel: +49 211 6792 160
bDepartment of Theoretical Chemistry, Max-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, D-45470 Mülheim an der Ruhr, Germany
cCenter for Electrochemical Sciences, Ruhr-Universität Bochum, Universitätstraße 150, D-44780 Bochum, Germany
dInstituto de Química Física “Rocasolano”, CSIC, C. Serrano 119, E-28006 Madrid, Spain
First published on 1st March 2013
Abstract
The impact of electrolyte constituents on the interaction of hydrogen peroxide with polycrystalline platinum is decisive for the understanding of the selectivity of the oxygen reduction reaction (ORR). Hydrodynamic voltammetry measurements show that while the hydrogen peroxide reduction (PRR) is diffusion-limited in perchlorate- or fluoride-containing solutions, kinetic limitations are introduced by the addition of more strongly adsorbing anions. The strength of the inhibition of the PRR increases in the order ClO4− ≈ F− < HSO4− < Cl− < Br− < I− as well as with the increase of the concentration of the strongly adsorbing anions. Electronic structure calculations indicate that the dissociation of H2O2 on Pt(111) is always possible, regardless of the coverage of spectator species. However, the adsorption of H2O2 becomes strongly endothermic at high coverage with adsorbing anions. A comparison of our observations on the inhibition of the PRR by spectators with previous studies on the selectivity of the ORR shows that oxygen is reduced to H2O2 only under conditions at which the PRR kinetics is significantly limited, while the ORR proceeds with a complete four-electron reduction only when the PRR is sufficiently fast. Therefore, only a H2O2-mediated pathway that includes a competition between the dissociation and the spectator coverage-dependent desorption of the H2O2 intermediate is enough to explain and unify all the observations that have been made so far on the selectivity of the ORR.
Introduction
The oxygen reduction reaction (ORR) is a fundamental reaction that plays an important role in many disciplines such as energy conversion, corrosion or biology.1–6 Even though the ORR has been extensively investigated in experimental and computational studies, its mechanism remains under discussion.7–13 In particular, the role of hydrogen peroxide in the reaction mechanism is still controversial. Apart from its fundamental importance, the understanding of the participation of H2O2 in the ORR is also significant from a technological point of view, since H2O2 is an undesirable product in low-temperature H2/air fuel cells having a detrimental impact on the fuel cell durability.14The reduction of oxygen to water involves four electron transfer steps (coupled or decoupled with proton transfer)15 and one O–O bond breaking step. Even though these elementary steps cannot occur simultaneously,16 they may take place very fast one after the other, which makes the distinction between them rather difficult. When an oxygen molecule is adsorbed on a metal surface, the cleavage of the O–O bond may occur prior to the first, second or third electron transfer step. The position of the bond-breaking step in the reaction sequence is typically used as the criterion to distinguish between the three proposed mechanisms of the ORR (visualized in Scheme 1):17–19 (i) the dissociative mechanism, in which the O–O bond breaking precedes all electron transfer steps (ii) the 1st associative mechanism, where one electron transfer step precedes the bond breaking and (iii) the 2nd associative (or “peroxo”-) mechanism, in which two electron transfer steps occur forming hydrogen peroxide before the bond breaks.
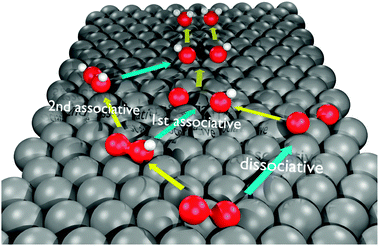 | ||
| Scheme 1 Proposed oxygen reduction reaction mechanisms. The yellow arrows indicate electron transfer steps; the azure arrows indicate O–O bond-breaking steps. | ||
None of the three mechanisms can be ruled out using quantum chemical calculations, since the energy barriers for the decisive steps are rather similar.19–21 In addition, the experimental exploration of the ORR mechanisms, for instance by in situ spectroscopic techniques, is not an easy task.9
The introduction of the rotating ring-disc electrode (RRDE) by Frumkin and Nekrasov22 enabled the probing of the amount of H2O2 that is formed during ORR and escapes from the vicinity of the working disc electrode, leading to a phenomenological distinction between two pathways (Scheme 2):23–25 (i) the “direct” (or “4-electron”) pathway, in which O2 is reduced to water without the detection of H2O2 on the ring of the RRDE, and (ii) the “series” (or “2-electron”) pathway, in which H2O2 is detected as an intermediate of the ORR.
 | ||
| Scheme 2 Proposed oxygen reduction reaction pathways. | ||
Due to its convenience, this phenomenological distinction between pathways is widely used nowadays. However, it must be kept in mind that this offers only a macroscopic description of the overall reaction, thus obscuring details of the reaction mechanism at the microscopic level. In particular, the detection of H2O2 is a proof that the 2nd associative mechanism is operative to a certain extent, as it is the only one that involves H2O2 formation. However, a lack of detection of H2O2 does not support or exclude any of the three mechanisms, as H2O2 may be formed at the interface but dissociate before it is transported away from the diffusion layer.
Hydrogen peroxide has been detected in the hydrogen adsorption region on Pt(111) and Pt(100) in weakly adsorbing electrolytes, as well as at more positive potentials (i.e. in the double-layer region) in solutions containing intentionally added anionic impurities (e.g., chloride) on any kind of Pt surface.26–34 An increased H2O2 formation (or a lower “effective” number of electrons transferred) has been additionally observed either when the loading of high-surface-area Pt catalysts is substantially decreased or when oxygen mass transport rates are very high (e.g., by the use of microelectrodes).7,12,35–42 These studies indicate that under these conditions, the ORR proceeds (at least in part) through the formation and desorption of H2O2, a fraction of the latter being re-adsorbed and eventually further reduced to water. Proving or disproving that the desorption–re-adsorption–dissociation scheme36,38 is universal for the ORR would certainly be a major milestone for the understanding of the ORR.
In order to elucidate the participation of H2O2 in the ORR, it is essential to understand the electrochemistry of H2O2 under conditions relevant to the ORR. However, the current understanding of the interaction of H2O2 with metal surfaces which is used to interpret observations during the ORR has evolved from old, poorly reproducible and eventually inconclusive data, as summarized in ref. 43. Identifying such shortcomings in the old literature, our group as well as other groups have recently reinvestigated the interaction of H2O2 with noble and non-noble metal catalysts.43–46 We previously showed that the kinetics of the hydrogen peroxide reduction and oxidation reactions (PROR) on Pt(poly) are very facile in electrolytes free from strongly adsorbing species.43,44 Depending on the electrode potential, either the hydrogen peroxide reduction reaction (PRR) to H2O or the hydrogen peroxide oxidation reaction (POR) to O2 occur very fast upon interaction with the Pt(poly). In such electrolytes, the total rate of H2O2 decomposition (given by the sum of the POR and PRR rates) is always controlled by mass transport of H2O2 due to the fast kinetics of both reactions on Pt(poly). In the following, we demonstrate how strongly adsorbed anionic spectator species can change the picture by influencing the interaction of hydrogen peroxide with Pt(poly). Finally, we correlate the findings of this paper on the H2O2 electrochemistry with previous RRDE measurements on the impact of such adsorbates on the ORR pathways.
Experimental section
Electrochemical measurements
The electrochemical measurements were conducted in a Teflon three-compartment electrochemical cell using a rotating disc electrode (RDE) setup with a Gamry Reference 600 potentiostat and rotation control (Radiometer Analytical). The volume of the electrolyte was 50 mL and the experiments were performed at room temperature. The potentiostat, the rotator and the gas flow were automatically controlled and programmed using an in-house-built LabVIEW software.47 The working electrode was a polycrystalline platinum disc of 5 mm diameter. The disc was embedded in a Teflon cylinder and polished with a silica suspension (final polishing suspension: 0.1 μm, Buehler; polishing cloth: Struers, MD Chem) prior to the measurements. A graphite rod and a saturated Ag/AgCl electrode (Metrohm), each separated by a Nafion membrane from the main cell compartment, served as the counter and the reference electrodes, respectively. All potentials in the paper are expressed with respect to the reversible hydrogen electrode (RHE) potential, which was determined versus the potential of the Ag/AgCl electrode in a hydrogen-saturated solution before each measurement. Positive feedback was used to compensate for the electrolyte resistance and the residual uncompensated resistance was less than 2 Ω in all experiments. The solutions were freshly prepared using ultrapure water (18 MΩ, TOC < 3 ppb, ELGA) and superpure chemicals (Merck, Suprapur®). The gases were provided by Air Liquide (class: 5.0 N).Computational details
For the quantum chemical investigations, a cluster ansatz was applied to model a Pt(111) surface using the bottom plane of a Pt37 cluster of hemispherical shape.48 Former investigations showed that small clusters with at least three layers of Pt and a diameter of approx. 1 Å yield results sufficiently close to those of slab calculations.19,49 The distance between the platinum atoms was the bulk distance of 2.78 Å.19 Furthermore, a ring of 12 water molecules was placed over the platinum atoms on the perimeter of the bottom plane to minimize the influence of edges as adsorption sites and to model the local environment of a water-covered surface. Seven Pt atoms are left free to adsorb H2O2 or chlorine atoms. Different surface coverages with spectator species were simulated by adsorbing up to three chlorine atoms at the surface.The calculations were carried out using the Orca program package50 at the RI-BP86/Def2-TZVP level of theory,51–53 including the Stuttgart pseudo potential (60 electrons) for platinum.54,55 More details for the quantum chemical calculations are available in the ESI.†
Results
Influence of (bi)sulfate adsorption
Fig. 1 shows the hydrodynamic voltammograms recorded in solutions of different concentrations of H2SO4, saturated with (a) argon (b) oxygen and (c) argon additionally containing 1 × 10−3 M H2O2. The corresponding voltammograms in 0.1 M HClO4 are also depicted for comparison (dotted curves). The voltammogram features of Pt single-crystal surfaces have already been described in detail in the literature.29,56–58 On the basis of these results, the current responses recorded for Pt(poly) in different solutions (Fig. 1a) can be interpreted, which will be useful for the following analysis of the ORR and PROR responses. In particular, the background response in sulfuric acid shows sharper peaks in the hydrogen adsorption region as compared to perchloric acid. This is a result of the adsorption–desorption of (bi)sulfate ions and the concomitant desorption–adsorption of hydrogen.29 Moreover, the area under the “oxide” reduction peak decreases and the onset potential for the adsorption of oxygenated species shifts more positively upon addition of sulfuric acid, both indicating the competition between the adsorption of oxygenated species and (bi)sulfate ions.57,58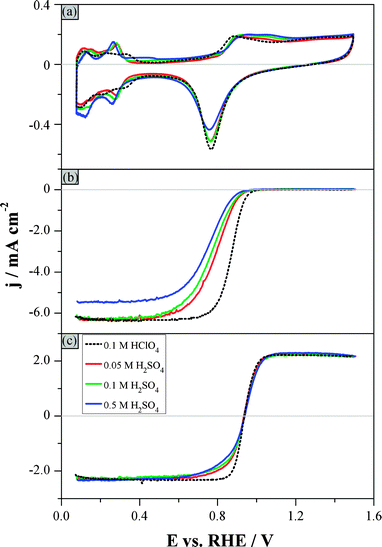 | ||
| Fig. 1 Hydrodynamic voltammograms in electrolytes of different H2SO4 concentrations and in HClO4 (see figure legend): (a) Ar-purged electrolyte, (b) O2-purged electrolyte, (c) Ar-purged electrolyte additionally containing 1 × 10−3 M H2O2. Rotation rate: 1600 rpm. Scan rate: 0.1 V s−1. Fig. 1b and c show only the positive-going, background-corrected sweeps. | ||
It is well known that the adsorption of (bi)sulfate ions causes an inhibition of the ORR on all low-index Pt single-crystal surfaces.29 This can also be seen in Fig. 1b, where the onset potential for the ORR shifts towards more negative potentials as the concentration of H2SO4 increases, while the potential region at which the current is controlled by both kinetics and mass transport (often called “mixed” region) is extended by approximately 0.2 V compared to 0.1 M HClO4. The kinetic current density for the ORR at +0.9 VRHE (SA0.9V) decreases in the order: 0.1 M HClO4 (2.0 mA cm−2) > 0.05 M H2SO4 (0.44 mA cm−2) > 0.1 M H2SO4 (0.34 mA cm−2) > 0.5 M H2SO4 (0.19 mA cm−2). Namely, the SA0.9V decreases by a factor of 4 from 0.1 M HClO4 to 0.05 M H2SO4, and by a factor of 2.5 when increasing the H2SO4 concentration from 0.05 M to 0.5 M. Note that the lower diffusion-limited current density in the 0.5 M H2SO4 solution compared to 0.1 M HClO4 is due to the decreased solubility of O2 in the former solution, while the difference in the viscosity (and thus the diffusion coefficient) is negligible.
The current for the H2O2 reduction is controlled by mass transport of H2O2 only up to +0.6 VRHE in all concentrations of sulfuric acid studied; this is around 0.25 V more negative compared to 0.1 M HClO4 (Fig. 1c). Above +0.6 VRHE the current in H2SO4 deviates from that in HClO4 leading to a broader “transition region” (as previously43,44 defined to describe the potential region between the diffusion-limited PRR and POR regions). This deviation is more pronounced for the more concentrated H2SO4 solutions. The sharp transition from negative to positive currents in the HClO4 electrolyte above +0.85 VRHE is due to the competition between the facile oxidation and reduction of H2O2.43 The observed deviation of the response in H2SO4 compared to that in HClO4 at potentials between +0.6 VRHE and +0.85 VRHE is related to the adsorption of (bi)sulfate ions and the consequent inhibition of the dissociative adsorption and reduction of H2O2 at potentials below the “oxide” formation. As the potential increases, adsorbed (bi)sulfate ions are gradually displaced by adsorbed oxygenated species,58 and the consequent decrease of the (bi)sulfate coverage allows the POR to occur eventually as fast as in HClO4.
If one takes into account the very high H2SO4 concentrations that were used, the influence of (bi)sulfate adsorption on the H2O2 reactions cannot be considered as very strong. Additionally, the increase of the concentration of H2SO4 by one order of magnitude does not significantly alter the current response in Fig. 1c, which means that the inhibition of H2O2 reactions due to (bi)sulfate adsorption cannot become severely stronger. Therefore, it can be concluded that even though (bi)sulfate adsorption causes some inhibition of the PRR in the potential region between +0.6 VRHE and +0.85 VRHE, the reaction can still proceed with a nearly diffusion-limited rate. It is worth mentioning that this is not the case for the ORR, where (bi)sulfate adsorption has a much more pronounced influence (see Fig. 1b). This difference in the strength of the effect of (bi)sulfate on the ORR and the PRR will be discussed below.
Influence of halide adsorption
Fig. 2 shows the hydrodynamic voltammograms recorded in 0.1 M HClO4 solutions with an increasing concentration of NaCl and saturated in (a) argon, (b) oxygen and (c) argon additionally containing 1 × 10−3 M H2O2. The background current peaks in the hydrogen adsorption–desorption region in Fig. 2a shift towards more negative potentials and become sharper with increasing chloride concentrations. These changes indicate that chloride adsorption–desorption and hydrogen desorption–adsorption occur concomitantly in the potential region below +0.3 VRHE.59–61 Increasing the potential above the potential of zero total charge during the positive-going sweep increases the chloride coverage,62 which in turn suppresses the adsorption of oxygenated species. Only at much more positive potentials Clad becomes replaced by oxygenated species.33,61,63 The suppression of the “oxide” formation due to chloride adsorption is also responsible for the decrease of the charge associated with the reduction of the irreversible “oxide”, as the chloride concentration increases (Fig. 2a).63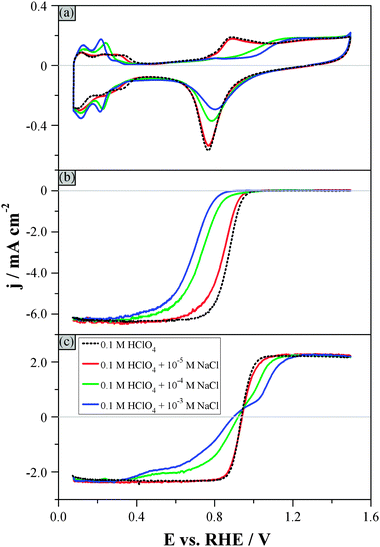 | ||
| Fig. 2 Hydrodynamic voltammograms in 0.1 M HClO4 electrolytes containing different concentrations of Cl− ions (see figure legend): (a) Ar-purged electrolyte, (b) O2-purged electrolyte, (c) Ar-purged electrolyte additionally containing 1 × 10−3 M H2O2. Rotation rate: 1600 rpm. Scan rate: 0.1 V s−1. Fig. 2b and c show only the positive-going, background-corrected sweeps. | ||
Chlorides are thus adsorbed on the Pt(poly) surface in the whole potential region between the hydrogen adsorption and the onset of adsorption of oxygenated species, and cause the inhibition of the ORR, as it is evidenced by the considerable shift of the onset potential for the ORR to higher overpotentials with the increase of the concentration of NaCl (Fig. 2b).33,64 In particular, the addition of 1 × 10−3 M Cl− in the 0.1 M HClO4 solution leads to a decrease by two orders of magnitude of the SA0.9V. Mass transport control of the reaction rate is achieved at much more negative potentials than in the chloride-“free” electrolyte. For instance, in the solution containing 1 × 10−3 M Cl− the diffusion-limited current is reached only below +0.3 VRHE.
Even though the presence of 1 × 10−5 M Cl− is already enough to influence the ORR, neither the PRR nor the POR is significantly affected by this concentration (Fig. 2c). However, higher chloride concentrations can severely inhibit the PRR: in a 1 × 10−4 M Cl− solution, the diffusion-limited current can be reached only in the hydrogen adsorption region, i.e. at potentials below +0.3 VRHE. Above +0.3 VRHE, the Clad coverage increases – as explained above – so the PRR is strongly suppressed and the negative current decreases, deviating from the diffusion-limited value, i.e. from the response in the chloride-free electrolyte. The deviation becomes more pronounced for the solutions containing higher chloride concentrations. A constant-potential electrolysis experiment (+0.75 VRHE) in a 0.1 M HClO4 + 1 × 10−3 M NaCl + 2 × 10−3 M H2O2 electrolyte confirmed that the H2O2 concentration decay deviates significantly from that expected for a diffusion-limited reaction (see Fig. S1, ESI†). Thus, in contrast to a chloride-“free” solution where the experimentally observed concentration decay is limited by the mass transport of H2O2 in the whole potential region studied,43 the decrease of the negative current at potentials above +0.3 VRHE is due to the inhibition of the PRR.
As the potential becomes more positive, the chloride coverage decreases due to the displacement of Clad by oxygenated species, therefore the inhibition of the H2O2 interaction with the surface weakens, similarly to what was described above for (bi)sulfate. The positive current increases rapidly and becomes eventually controlled by H2O2 diffusion at around +1.2 VRHE for the 1 × 10−3 M Cl− solution (Fig. 2c). A more positive potential is required to reach the diffusion-limited rate of the POR for higher concentrations of chloride in solution, as the latter leads to enhanced Clad coverage up to that potential, in agreement with the positively shifted adsorption of oxygenated species in Fig. 2a.
Fig. 3 shows the hydrodynamic voltammograms recorded in 0.1 M HClO4 solutions containing 1 × 10−4 M of NaX, where X: F−, Cl− or Br−. The influence of the halides on the background responses (Fig. 3a) becomes stronger following the order F− < Cl− < Br− (Fig. 3a), reflecting the increasing halide coverage on platinum for identical concentrations.65 In fact, a 10−4 M concentration of fluoride in the HClO4 solution is low enough to have practically no effect on the background responses. This is because the adsorption strength of fluoride and perchlorate ions, and thus their influence on the adsorption of hydrogen and of oxygenated species, is about the same.60,65 In contrast, the same concentration of chloride and bromide significantly suppresses both processes: (i) the adsorption of oxygenated species, as can be seen by the positive shift of the onset potential in the positive-going sweep, and (ii) the hydrogen adsorption, as can be seen by the negative shift of the onset potential for Hupd adsorption in the negative-going sweep. Both shifts increase with the size of the halide ions (see Fig. 3a).61
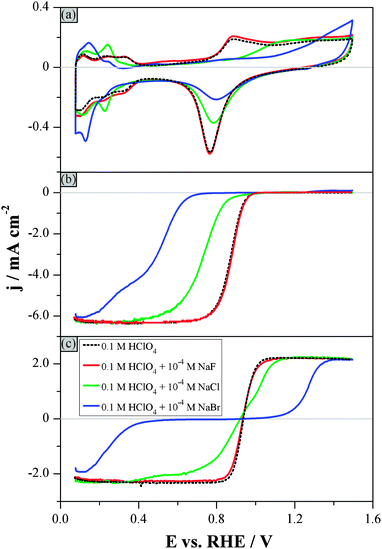 | ||
| Fig. 3 Hydrodynamic voltammograms in 0.1 M HClO4 electrolytes containing 1 × 10−4 M NaX where X: F−, Cl− or Br− (see figure legend): (a) Ar-purged electrolyte, (b) O2-purged electrolyte, and (c) Ar-purged electrolyte additionally containing 1 × 10−3 M H2O2. Rotation rate: 1600 rpm. Scan rate: 0.1 V s−1. Only the background-corrected, positive direction of the sweeps is shown in Fig. 3b and c. | ||
While the oxygen reduction reaction is not influenced by the adsorption of fluoride (Fig. 3b), a considerable shift of the onset potential for the ORR is observed in the presence of chloride and bromide, resulting in a stronger inhibition of oxygen reduction. In the bromide-containing solution, the ORR commences at around +0.6 VRHE, which is approximately 0.35 V more negative than the onset potential in the presence of the same concentration of fluoride. It should be noted that the inflection point in the curve corresponding to the ORR in the bromide-containing solution, which signals the switch of the ORR from a 2-electron to a 4-electron reduction, coincides with the onset of the PRR.
The same trend with respect to the different anions is qualitatively followed for the hydrogen peroxide reactions (Fig. 3c). Again, the interaction of H2O2 with platinum in the presence of fluoride is practically unaffected and comparable to that in pure HClO4. The significantly stronger inhibition of H2O2 reactions by Clad has been described above, while Brad has an even stronger effect. Indeed, the adsorption of bromide is so strong that it suppresses completely the H2O2 reactions in the potential region between +0.4 VRHE and +1.1 VRHE. This implies that neither the PRR nor the POR are possible in this potential region. The POR commences only above +1.1 VRHE due to the displacement of Brad by oxygenated species, and the current increases gradually, reaching the diffusion-limited value at +1.3 VRHE. The good agreement in all halide-containing solutions between the potentials at which the POR starts (Fig. 3c) and at which the displacement of halides by oxygenated species takes place (Fig. 3a) is an additional indication that the inhibition of POR is caused by the adsorbed halides. On the other hand, the PRR in the bromide-containing electrolyte starts only below +0.4 VRHE, which is 0.2 V more negative than the onset potential for ORR in the same solution.
It is noteworthy that, while Clad hinders the ORR more strongly than the PRR, Brad influences the two-electron reduction of O2 to a lesser extent than the further reduction of H2O2 to H2O. A similar effect was observed in an additional experiment in the presence of iodide (0.1 M HClO4 + 1 × 10−4 M NaI; see Fig. S2, ESI†): the PRR was completely suppressed in the potential region studied, while the two-electron reduction of oxygen could take place at potentials below +0.4 VRHE. This behaviour will be discussed in the Discussion section.
Electronic structure calculations
Electronic structure calculations were carried out in order to obtain trends of the atomistic details that give rise to the major influences of co-adsorbed species on the reactions discussed above. For this purpose, three model surfaces were used to resemble the local environment of a given reacting species on the surface. While these systems are far from a detailed model that includes all relevant parameters that influence reactivity, they still allow assessing whether adsorbates influence the adsorption and/or bond breaking due to local electronic or steric effects.In a previous paper,43 we showed that H2O2 dissociates into two OHad with a barrier of 0.22 eV, when adsorbed on a bare Pt(111) surface (an adsorption energy of −0.34 eV), modeled by a cluster. The dissociation energy is −1.55 eV (see Fig. 4a). Experimental evidence for the dissociative adsorption of H2O2 on Pt has been given previously using surface-enhanced Raman spectroscopy.66 If two of the seven platinum atoms are covered by chlorine atoms, the adsorption energy of H2O2 out of the gas phase remains exothermic (−0.37 eV), as shown in Fig. 4b. Taking the solvation energy of H2O2 into account (literature values range from −0.3 eV to −0.6 eV as indicated by a grey bar in Fig. 4), the tendency for H2O2 to adsorb is small, both on the clean Pt(111) surface and on a Pt(111) surface with two chlorine atoms adsorbed. It should be noted, however, that interactions of the adsorbed H2O2 with the solvent were neglected, so this is just a lower limit for the adsorption energy. Taking solvation effects of the adsorbed H2O2 into account, a further stabilization and thus a further increase of the adsorption energy must be expected. The barrier for H2O2 dissociation for this relatively low coverage with chloride is 0.14 eV, which is similar to that for the chlorine-free surface (the slight difference between the two barriers is due to Cl–H hydrogen bonding, see ESI†). The dissociation is still highly exothermic, with a dissociation energy of −1.40 eV. Hence, the results of the simulations indicate that the presence of up to two chlorine atoms does not introduce major differences in the local reactivity of H2O2 on the surface.
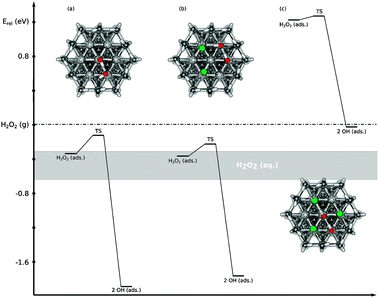 | ||
| Fig. 4 Energy diagram for the dissociation of H2O2 on the Pt(111) surface model with (a) no, (b) two and (c) three adsorbed chlorine atoms. The energies are given relative to H2O2 in the gas phase (dashed line). The grey bar corresponds to H2O2 in solution (see the text). | ||
In order to estimate whether a situation exists in which steric hindrance might prevent dissociation of H2O2 by O–O bond cleavage, a model for an extreme situation was chosen. In this model, three out of the seven Pt atoms of the surface were covered by chlorine atoms. Under these conditions, the distance between two chlorine atoms is 3.8 Å, which is very close to the sum of their van der Walls radii (3.5 Å),67 thus leaving only minimal space for further adsorbates (see Fig. S3, ESI†). As a consequence, the adsorption of H2O2 at a platinum atom of this model is not favorable energetically. For a structure including H2O2 adsorption, the distance between the chlorine atoms increases to 5.1 Å (see Fig. S5, ESI†), while the sum of the van der Waals radii of the two chlorine atoms and the diameter of the oxygen atom (that is located between two chlorine atoms) is 6.3 Å. However, the barrier to dissociate H2O2 under these circumstances is only 0.05 eV and the dissociation energy is −1.25 eV. As these values are of comparable magnitude to the results obtained on the other models, one can assume that irrespective of local spatial restrictions due to high chlorine coverage, the kinetics of H2O2 dissociation on the surface should be more or less the same. While detailed and accurate data for adsorption/solvation equilibria would be required for a more complete description, the limited results from this study allow at least presuming that the inhibition of the PRR at high chlorine coverages might be due to an inhibited adsorption of H2O2, rather than due to an impeded dissociation.
Turning to the behavior of oxygen, it first needs to be noted that adsorption of an oxygen molecule is always more favorable than that of a H2O2 molecule, due to the poorer stabilization of O2 in aqueous solution. The dissociation of O2 at a bare Pt(111) surface model (see Fig. 5a) proceeds with a barrier of +0.72 eV and a dissociation energy of −0.53 eV, in agreement with values obtained by Jacob and Goddard.19 When two of the seven Pt atoms are covered with chlorine atoms, breaking the O–O bond of oxygen becomes highly endothermic (+1.01 eV). Therefore, the dissociation of O2 in this model system is inhibited by the local environment, in contrast to H2O2 dissociation that remains unaffected in the same model, as described above. This contrast is caused by the different binding modes preferred by the products, Oad in the case of O2 and OHad in the case of H2O2: while OHad preferentially binds on top of a platinum atom (like Clad) and hence just one platinum atom is required, atomic oxygen preferentially adsorbs on an fcc-site, which requires three platinum atoms.68,69 For O2 dissociation on the model system in Fig. 5b, the nearby chlorine atoms restrict the two oxygen atoms to adsorb on top sites, accompanied by a drastic increase of relative energy of this product structure (compare relative energies for the products of O2 dissociation in Fig. 5a and b). In this context, it is interesting to note that Eikerling and co-workers calculated binding energies of atomic oxygen for different binding sites on Pt nanoparticles and reported values of approximately −0.85 eV for fcc, −0.50 eV for hcp, −0.38 eV for bridge and 0.41 eV for top sites.48 This strong decrease of oxygen binding strength from fcc to top sites indicates that the unfavorable O2 dissociation in the presence of chlorine atoms mostly stems from the spatial restrictions imposed by the co-adsorbates, preventing the product Oad to adsorb in the preferred fcc hollow sites.
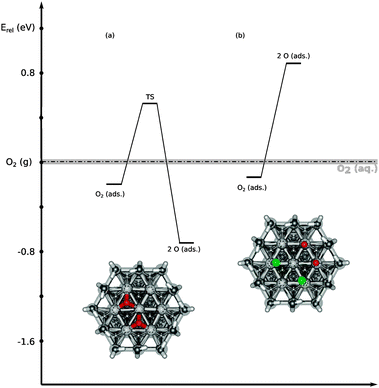 | ||
| Fig. 5 Energy diagram for the dissociation of O2 on the Pt(111) surface model with (a) no and (b) two adsorbed chlorine atoms. The energies are given relative to O2 in the gas phase (dashed line). | ||
Discussion
Correlation between the PRR and the ORR
In a previous paper, we showed that the kinetics of both H2O2 reactions, the PRR at low potentials and the POR at high potentials, are very fast in the absence of inhibiting spectators in a highly pure, weakly adsorbing electrolyte (0.1 M HClO4).43 The coverage with inhibiting spectator species is negligible in this case in the potential region between +0.2 VRHE and +1.5 VRHE.‡ As a consequence, the H2O2 decomposition occurs with a total rate that is controlled only by the mass transport of H2O2 in the whole potential window between +0.2 VRHE and +1.5 VRHE. This translates into an infinitesimally low concentration of H2O2 at the electrode–electrolyte interface. In particular, due to the fast kinetics of the reactions of H2O2, the local concentration of hydrogen peroxide must be close to a limit imposed by thermodynamics, which equals 8.67 × 10−19 M.43The results presented in this paper show that the PRR and to a lesser extent the POR are slowed down when the coverage with inhibiting species increases. The extent of the inhibiting effect is related to the surface coverage with such spectators. The electronic structure calculations indicate that the observed inhibition of the PRR at high coverage is related to an inhibition of the adsorption of H2O2. Depending on the coverage with inhibiting spectator species, the total rate of H2O2 decomposition will eventually be limited by the slow kinetics of the PRR and the POR (rather than by the mass transport of H2O2, as for the weakly adsorbing electrolytes). As a consequence, considerable local H2O2 concentrations are expected at the vicinity of the electrode surface.
Based on this difference between weakly and strongly adsorbing electrolytes, we will summarize and discuss below our observations for the PRR in relation to previous RRDE investigations for the selectivity of the ORR.
Two important conclusions can be drawn from the above discussion. First, there is a clear correlation between the inhibition of the PRR and the selectivity of the ORR towards the complete reduction to H2O. In particular, H2O2 formation during the ORR has been observed only under conditions in which the PRR is retarded, while the complete reduction of O2 to H2O on Pt is only observed under conditions in which H2O2 reduction to H2O is very fast. This consistency between previous RRDE investigations of the ORR and our observations made for the PRR suggests that the facile reduction of H2O2 to H2O is a prerequisite for the complete reduction of O2 to H2O, under the same conditions. Even though this can be so far stated only for platinum, literature data on other noble and non-noble materials,74–78 as well as preliminary results that we have obtained on Au(poly) and Rh(poly), seem to confirm this concept. This strongly implies an important role of H2O2 in the oxygen reduction reaction, as it will be also discussed below.
The second important conclusion is that the nature of the inhibiting effect that the spectators cause on the PRR (and on the ORR selectivity) is the same, regardless of the nature of the spectators. The effect becomes stronger for higher surface coverage, which can be reached either by increasing the strength of the adsorption (increasing from fluoride, where an effect can hardly be seen, to (bi)sulfate, chloride, bromide, and finally to iodide), or by increasing the anion concentration. The relevance of the surface coverage and the apparent insensitivity to the chemical nature of the adsorbed anions suggest that the latter act mainly as blocking species, and that the inhibition of the PRR is essentially due to an atomic-ensemble effect,79 rather than to an alteration of the electronic structure of the Pt atoms that remain exposed to the solution (electronic effect). As shown by the theoretical calculations above, the spatial restrictions imposed by spectators on the adsorption of reactants, intermediates and products affect their corresponding adsorption energies.
Mechanistic insight into the ORR – desorption versus dissociation
The mechanistic details of the role of H2O2 in the oxygen reduction, and of the observed changes in the selectivity during the ORR, are not yet fully elucidated. However, the findings of this work regarding the impact of spectators on the interaction of H2O2 with platinum and the discussion made above converge to a plausible scheme for the ORR that can describe all the macroscopic observations that have been made so far for the ORR selectivity, as follows.The electronic structure calculations presented in this paper, as well as the findings in ref. 43, show that platinum is intrinsically a good catalyst for the dissociation of H2O2. In particular, the barrier for breaking the O–O bond of adsorbed H2O2 is independent of the local environment and is consistently very low at all spectator coverages. In contrast, the calculations indicate that spectators affect strongly the adsorption of H2O2, which is slightly exothermic at low to medium coverage, but becomes highly endothermic at high coverage. Once adsorbed H2O2 is formed during the ORR, a competition between (spectator coverage-dependent) desorption and (spectator coverage-independent) dissociation will occur. The desorbed H2O2 may experience an additional competition between re-adsorption and diffusion away from the electrode surface.
At low spectator coverage, H2O2 desorption is not very favorable and it is very likely that it does not supersede the very facile H2O2 dissociation. Even if some H2O2 desorbs (note that this process is slightly endothermic at low spectator coverage) re-adsorption is still very probable, unless the mass transport of the produced H2O2 is very fast. Therefore, at low spectator coverage, H2O2 cannot accumulate on the surface and exceed the infinitesimal equilibrium concentration imposed by thermodynamics (8.67 × 10−19 M).
At high spectator coverage, the H2O2 desorption–adsorption equilibrium changes and now desorption of H2O2 is very favorable, competing strongly (or even dominating) over dissociation, while re-adsorption becomes endothermic. Thus, there is a high probability that the formed H2O2 will desorb rather than dissociate and H2O2 will be detected in the electrolyte as long as it diffuses away before re-adsorbing. Partial re-adsorption of the desorbed H2O2 will be possible only at spectator coverages at which H2O2 adsorption is only slightly endothermic.
Interestingly, the effect described above for high coverage of spectators was reported even in weakly adsorbing electrolytes in previous papers mainly by the groups of Kucernak7 and Behm.36 The H2O2 produced upon increasing mass transport rates (e.g. by the use of ultramicroelectrodes) was attributed to a higher probability for H2O2 to escape prior to re-adsorption, which cannot be observed under the slow (compared to ultramicroelectrodes) mass transport conditions of an RRDE. It needs to be noted, however, that only under equilibrium conditions, the spectator coverage (at a given surface) depends on the nature and the concentration of the electrolyte ions as well as on the electrode potential. In contrast, under non-equilibrium conditions (e.g. during potentiodynamic experiments and low anionic concentration, like in the current and the previous studies cited above) the spectator coverage depends additionally on the mass transport of the inhibiting ions from the solution to the surface at a given time, and thus does not necessarily equal the equilibrium spectator coverage for the corresponding potential. Therefore, it is not only the mass transport rate of the produced H2O2 away from the surface that is enhanced, e.g. for the case of ultramicroelectrodes, but also the mass transport rate of trace impurities towards the surface, as also suggested previously.80 In principle, the increase of the mass transport rates of both H2O2 and chloride cannot be decoupled in these experiments, and will eventually lead to a higher coverage of chloride than the one expected for the slow mass transport conditions of an RRDE, even for the highest purity weakly adsorbing electrolyte. Under these conditions, and in agreement with the case of high spectator coverages described above, the H2O2 desorption and release will therefore be stimulated at high mass transport rates of both H2O2 and chloride. Obviously, the re-adsorption of H2O2 prior to diffusion may still be possible as described in the desorption/re-adsorption/reaction model,7,36 but only in case that the H2O2 adsorption is still only slightly endothermic at the given spectator coverage.
Therefore, only one H2O2-mediated ORR pathway (equivalent to the “series” pathway) is enough to explain and unify all the observations that have been made so far for the ORR selectivity. In this scheme (Scheme 3), the decisive step on whether H2O2 or H2O will be formed is the competition between dissociation and desorption of the produced H2O2 intermediate, which is related to the surface and/or interface structure. For surfaces that are excellent catalysts for the dissociation of H2O2, as for example platinum, this competition depends strongly on the presence of adsorbates that can stimulate H2O2 desorption and make it capable of competing dissociation only at high spectator coverages. Hence, the “direct” pathway can be eliminated as it is only a limiting case in which the dissociation is much faster compared to desorption. It has to be emphasized, however, that this is still not sufficient proof that the ORR always and solely proceeds through the 2nd associative mechanism, and hence to exclude the other two mechanisms.
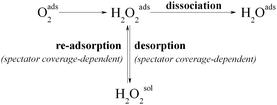 | ||
| Scheme 3 A single H2O2-mediated pathway that includes a competition between dissociation and desorption of H2O2 as the decisive step for the ORR selectivity. | ||
Conclusions
We will summarize below the main findings of this work:• Hydrodynamic voltammetry studies of the PRR on Pt(poly) in the presence of adsorbing anions show an increasingly inhibiting effect along the series ClO4− ≈ F− < HSO4− < Cl− < Br− < I− and with the increase of the concentration, introducing kinetic limitations on the PRR rate. At concentrations of 10−4 M, chloride hinders the ORR more than the PRR, while bromide and iodide block the PRR for a wider range of potentials than the ORR.
• Electronic structure calculations show that once hydrogen peroxide is adsorbed on Pt(111), it can dissociate with a low barrier at all chloride coverages. On the other hand, the adsorption of H2O2 becomes strongly endothermic (and desorption highly exothermic) at high chloride coverage.
• Under conditions in which the PRR is severely kinetically limited, the oxygen reduction forms hydrogen peroxide. However, when the PRR rate is limited by mass transport only, ORR proceeds with a complete 4-electron reduction. This coincidence strongly suggests that the feasibility of H2O2 reduction is a decisive prerequisite for the complete reduction of oxygen to water.
• The oxygen molecules that are reduced to hydrogen peroxide may either desorb or dissociate and further be reduced to water. The competition between the dissociation of H2O2 and the spectator coverage-dependent desorption of H2O2 is decisive for the selectivity of the ORR towards peroxide or water: at low coverage of inhibiting species, the desorption–adsorption equilibrium favors the adsorbed state while at high anion coverage, desorption of H2O2 becomes exothermic and basically irreversible.
Overall, the considerations made in this work have very important implications for the selectivity of the ORR and for the change in the reaction pathways for Pt-based materials often mentioned in the literature. In fact, only a H2O2-mediated pathway that includes a competition between the dissociation and the spectator coverage-dependent desorption of the H2O2 intermediate is enough to explain and unify all the observations that have been made so far on the selectivity of the ORR. The “direct” pathway is indeed a limiting case of the “series” pathway which arises when the rate of desorption of the formed H2O2 is sufficiently low compared to that of the H2O2 dissociation and further reduction. Previous macroscopic observations on the ORR selectivity using the RRDE, that have been interpreted as a suppression of the ORR without a change in the reaction pathway (e.g. for (bi)sulfate) or with a change in the reaction pathway (e.g. for Hupd, chloride or bromide), are only phenomenological and do not originate from an intrinsic property of platinum that determines the selectivity of the reaction. Instead, such macroscopically observed changes in the reaction selectivity are related to the impact of the adsorbed spectators on the competition between H2O2 desorption and dissociation.
Acknowledgements
J.C.M. acknowledges financial support by the Kekulé Fellowship from the Fonds der Chemischen Industrie (FCI). W.B.S. acknowledges funding from the Center for Electrochemical Sciences, Bochum.Notes and references
- M. K. F. Wikstrom, Nature, 1977, 266, 271–273 CAS.
- M. Stratmann and J. Muller, Corros. Sci., 1994, 36, 327–359 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS.
- Y.-C. Lu, H. A. Gasteiger and Y. Shao-Horn, J. Am. Chem. Soc., 2011, 133, 19048–19051 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS.
- S. L. Chen and A. Kucernak, J. Phys. Chem. B, 2004, 108, 3262–3276 CrossRef CAS.
- Y. X. Wang and P. B. Balbuena, J. Phys. Chem. B, 2005, 109, 14896–14907 CrossRef CAS.
- M. Shao, P. Liu and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 7408–7409 CrossRef CAS.
- M. J. Janik, C. D. Taylor and M. Neurock, J. Electrochem. Soc., 2009, 156, B126–B135 CrossRef CAS.
- V. Tripković, E. Skúlason, S. Siahrostami, J. K. Nørskov and J. Rossmeisl, Electrochim. Acta, 2010, 55, 7975–7981 CrossRef CAS.
- P. S. Ruvinskiy, A. Bonnefont, C. Pham-Huu and E. R. Savinova, Langmuir, 2011, 27, 9018–9027 CrossRef CAS.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 2948–2951 Search PubMed.
- N. Ramaswamy, N. Hakim and S. Mukerjee, Electrochim. Acta, 2008, 53, 3279–3295 CrossRef CAS.
- M. T. M. Koper, Phys. Chem. Chem. Phys., 2013, 15, 1399 RSC.
- M. T. M. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
- A. U. Nilekar and M. Mavrikakis, Surf. Sci., 2008, 602, L89–L94 CrossRef CAS.
- T. Jacob and W. A. Goddard, ChemPhysChem, 2006, 7, 992–1005 CrossRef CAS.
- J. A. Keith and T. Jacob, Angew. Chem., Int. Ed., 2010, 49, 9521–9525 CrossRef CAS.
- J. A. Keith, G. Jerkiewicz and T. Jacob, ChemPhysChem, 2010, 11, 2779–2794 CrossRef CAS.
- A. N. Frumkin and L. N. Nekrasov, Dokl. Akad. Nauk SSSR, 1959, 126, 115–118.
- H. S. Wroblowa, Y. C. Pan and G. Razumney, J. Electroanal. Chem., 1976, 69, 195–201 CrossRef CAS.
- A. J. Appleby and M. Savy, J. Electroanal. Chem., 1978, 92, 15–30 Search PubMed.
- J. C. Huang, R. K. Sen and E. Yeager, J. Electrochem. Soc., 1979, 126, 786–792 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1995, 99, 3411–3415 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1996, 100, 6715–6721 CrossRef.
- B. N. Grgur, N. M. Markovic and P. N. Ross, Can. J. Chem., 1997, 75, 1465–1471 CAS.
- N. Markovic, H. Gasteiger and P. N. Ross, J. Electrochem. Soc., 1997, 144, 1591–1597 CAS.
- N. M. Markovic and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117–229 CrossRef CAS.
- A. Damjanovic, M. A. Genshaw and J. O. M. Bockris, J. Electrochem. Soc., 1967, 114, 466–472 CAS.
- N. M. Markovic, H. A. Gasteiger, B. N. Grgur and P. N. Ross, J. Electroanal. Chem., 1999, 467, 157–163 CrossRef CAS.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- V. Stamenkovic, N. M. Markovic and P. N. Ross, J. Electroanal. Chem., 2001, 500, 44–51 CrossRef CAS.
- D. Pletcher and S. Sotiropoulos, J. Electroanal. Chem., 1993, 356, 109–119 CrossRef CAS.
- A. Schneider, L. Colmenares, Y. E. Seidel, Z. Jusys, B. Wickman, B. Kasemo and R. J. Behm, Phys. Chem. Chem. Phys., 2008, 10, 1931–1943 RSC.
- Y. E. Seidel, A. Schneider, Z. Jusys, B. Wickman, B. Kasemo and R. J. Behm, Faraday Discuss., 2008, 140, 167–184 Search PubMed.
- J. Fuhrmann, H. Zhao, H. Langmach, Y. E. Seidel, Z. Jusys and R. J. Behm, Fuel Cells, 2011, 11, 501–510 Search PubMed.
- K. Ke, T. Hatanaka and Y. Morimoto, Electrochim. Acta, 2011, 56, 2098–2104 CrossRef CAS.
- P. S. Ruvinskiy, A. Bonnefont and E. R. Savinova, Electrocatalysis, 2011, 2, 123–133 Search PubMed.
- M. Inaba, H. Yamada, J. Tokunaga and A. Tasaka, Electrochem. Solid-State Lett., 2004, 7, A474–A476 CrossRef CAS.
- A. Bonakdarpour, T. R. Dahn, R. T. Atanasoski, M. K. Debe and J. R. Dahn, Electrochem. Solid-State Lett., 2008, 11, B208–B211 CrossRef CAS.
- I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. U. Biedermann, A. A. Auer and K. J. J. Mayrhofer, Phys. Chem. Chem. Phys., 2012, 14, 7384–7391 RSC.
- I. Katsounaros and K. J. J. Mayrhofer, Chem. Commun., 2012, 48, 6660–6662 RSC.
- A. M. Gómez-Marín, K. J. P. Schouten, M. T. M. Koper and J. M. Feliu, Electrochem. Commun., 2012, 22, 153–156 Search PubMed.
- F. Jaouen and J.-P. Dodelet, J. Phys. Chem. C, 2009, 113, 15422–15432 CrossRef CAS.
- A. A. Topalov, I. Katsounaros, J. C. Meier, S. O. Klemm and K. J. J. Mayrhofer, Rev. Sci. Instrum., 2011, 82, 114103 CrossRef.
- L. Wang, A. Roudgar and M. Eikerling, J. Phys. Chem. C, 2009, 113, 17989–17996 CrossRef CAS.
- M. Kettner, W. B. Schneider and A. A. Auer, J. Phys. Chem. C, 2012, 116, 15432–15438 Search PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- K. Al-Jaaf-Golze, D. M. Kolb and D. Scherson, J. Electroanal. Chem., 1986, 200, 353–362 CrossRef.
- V. Stamenkovic, K. C. Chou, G. A. Somorjai, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2005, 109, 678–680 CrossRef CAS.
- A. Lachenwitzer, N. Li and J. Lipkowski, J. Electroanal. Chem., 2002, 532, 85–98 CrossRef CAS.
- N. Markovic, M. Hanson, G. McDougall and E. Yeager, J. Electroanal. Chem., 1986, 214, 555–566 CrossRef CAS.
- D. V. Tripkovic, D. Strmcnik, D. van der Vliet, V. Stamenkovic and N. M. Markovic, Faraday Discuss., 2009, 140, 25–40 RSC.
- M. W. Breiter, Electrochim. Acta, 1962, 8, 925–935 Search PubMed.
- N. Li and J. Lipkowski, J. Electroanal. Chem., 2000, 491, 95–102 CrossRef CAS.
- D. M. Novak and B. E. Conway, J. Chem. Soc., Faraday Trans., 1981, 77, 2341–2359 Search PubMed.
- T. M. Arruda, B. Shyam, J. M. Ziegelbauer, S. Mukerjee and D. E. Ramaker, J. Phys. Chem. C, 2008, 112, 18087–18097 CrossRef CAS.
- Y.-C. Chiu and M. A. Genshaw, J. Phys. Chem., 1969, 73, 3571–3577 CrossRef CAS.
- X. Li, D. Heryadi and A. A. Gewirth, Langmuir, 2005, 21, 9251–9259 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- A. Bogicevic, J. Strömquist and B. Lundqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, R4289–R4292 Search PubMed.
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717–4725 CrossRef CAS.
- E. Kirsten, G. Parschau, W. Stocker and K. H. Rieder, Surf. Sci. Lett., 1990, 231, L183–L188 Search PubMed.
- H. Ogasawara and M. Ito, Chem. Phys. Lett., 1994, 221, 213–218 CrossRef CAS.
- A. Kolics and A. Wieckowski, J. Phys. Chem. B, 2001, 105, 2588–2595 CrossRef CAS.
- E. Herrero, J. Mostany, J. M. Feliu and J. Lipkowski, J. Electroanal. Chem., 2002, 534, 79–89 CrossRef CAS.
- R. R. Adzić, N. M. Marković and V. B. Vešović, J. Electroanal. Chem., 1984, 165, 105–120 CrossRef CAS.
- R. Zeis, T. Lei, K. Sieradzki, J. Snyder and J. Erlebacher, J. Catal., 2008, 253, 132–138 CrossRef CAS.
- J. S. Jirkovský, M. Halasa and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2010, 12, 8042–8053 RSC.
- N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015–18026 CrossRef CAS.
- M. Campos, W. Siriwatcharapiboon, R. J. Potter and S. L. Horswell, Catal. Today, 2013, 202, 135–143 Search PubMed.
- A. Cuesta, ChemPhysChem, 2011, 12, 2375–2385 CrossRef CAS.
- D. A. Scherson and Y. V. Tolmachev, Electrochem. Solid-State Lett., 2010, 13, F1–F2 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed information about the steady-state electrolysis experiment in a chloride-containing electrolyte, the impact of iodide on ORR and PRR, and details of the electronic structure calculations. See DOI: 10.1039/c3cp50649e |
| ‡ Oxygenated species adsorbed above ca. +0.85 VRHE are not detrimental for the POR, and therefore not considered as “inhibiting” species, in contrast to the ORR. |
| This journal is © the Owner Societies 2013 |