 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of 1 mmol dm−3 concentrations of small molecules containing nitrogen-based cationic groups on the oxygen reduction reaction on polycrystalline platinum in aqueous KOH (1 mol dm−3)†
Ai Lien Ong*, Daniel K. Whelligan, Michael L. Fox and John R. Varcoe
Department of Chemistry, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford, GU2 7XH UK. E-mail: a.ong@surrey.ac.uk; Tel: +44 1483 682616
First published on 23rd September 2013
Abstract
Alkaline anion-exchange membranes (AAEMs) containing cationic head-groups (e.g. involving quaternary ammonium and imidazolium groups) are of interest with regard to application in alkaline polymer electrolyte fuel cells (APEFCs). This initial ex situ study evaluated the effect of 1 mmol dm−3 concentrations of model molecules containing (AAEM-relevant) cationic groups on the oxygen reduction reaction on a polycrystalline platinum disk (Ptpc) electrode in aqueous KOH (1 mol dm−3). The cationic molecules studied were tetramethylammonium (TMA), benzyltrimethylammonium (BTMA), 1-benzyl-3-methylimidazolium (BMI), 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane (BAABCO) and 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium (BOTMHA). Both cyclic and hydrodynamic linear sweep rotating disk electrode voltammetry techniques were used. The resulting voltammograms, derived estimates of apparent electrochemically active surface areas, Tafel slopes, apparent exchange-current densities and the number of electrons transferred (per O2 molecule) were compared. The results strongly suggest that 1 mmol dm−3 concentrations of BTMA, BAABCO, and (especially) BMI seriously inhibit the catalytic activities of Ptpc in an aqueous KOH electrolyte at 25 °C. The negative influence of (benzene-ring-free) TMA and Cl− anions (KCl control experiment) appeared to be less severe. The separation of the trimethylammonium group from the benzene ring via a hexyloxy spacer chain (in BOTMHA) also produced a milder negative effect.
Introduction
Alkaline anion-exchange membranes (AAEMs) are rapid developing core components in alkaline polymer electrolyte fuel cell (APEFC) technologies due to the perceived advantages of: (a) inhibiting CO2-derived precipitate formation during fuel cell operation; (b) the ability to use cheaper electrocatalysts; (c) improved oxygen reduction reaction (ORR) electrokinetics at the cathode (although hydrogen oxidation reaction [HOR] electrokinetics at the anode are poorer at high pH);1 (d) potential for minimised fuel cross-over (in non-H2 fuel cells); and (e) novel water management possibilities.2 Well-known research and development challenges for such fuel cell development focus on enhancing long-term component durabilities and reducing the cost-derived barriers to commercialisation. In this regard, researchers worldwide are searching for more stable AAEM macromolecular backbones and head-groups3 as well as investigating AAEM degradation pathways,4 how to enhance AEM head-group conductivities,5 the causes of AAEM head-group degradation,2a,d,5e,6 ways to reduce Pt loadings7 and Pt poisoning,8 alternative non-noble metal catalysts with acceptable performance,9 and new material design and engineering concepts.10,11Anion-exchange membranes (AEMs) are ion-exchange membranes containing fixed cationic sites attached to a macromolecular backbone; such membranes primarily conduct anions and transport water.5e The most commonly studied cationic sites are simple quaternary ammonium types but alternatives such as imidazolium-types and those created using the diamine 1,4-diazabicyclo[2.2.2]octane (DABCO) are also commonly encountered. Debates on the most promising AEM head-groups are routinely encountered. For instance, Qiu and co-workers5a have argued that imidazolium-functionalized membranes showed excellent chemical stability in alkali, compared to quaternary ammonium types, without significant loss of ion conductivities. Guo and colleagues5h also prepared imidazolium-AEMs that exhibited superior stabilities, both chemically and thermally, compared to quaternary ammonium functionalized AEMs with hydroxyl ion conductivities of 10−2 S cm−1. Similar observations were reported in the works by Ran et al.5i and Yan et al.5j However, Deavin et al.2a provided strong spectroscopic evidence that the performance of radiation-grafted benzylmethylimidazolium-containing AEMs exhibited significantly poorer chemical stabilities, compared to quaternary ammonium benchmark head-groups, under strong alkaline conditions.
The influence of the AEM polymer backbone and head-group chemistries on actual fuel cell performances can be diverse: for example, Li et al.5c have suggested that side-chain quaternary ammonium functional substituents in AEMs can enhance hydroxide conductivities and mitigate against excessive water swelling. However, the impact of the cationic head-group chemistries on the actual performances of the electrocatalysts has not, to date, been comprehensively studied. The presence of traces of these cationic head-groups can have a highly incapacitating effect towards the exceedingly sensitive electro-active surfaces of the catalysts.
Given the increasing market prices and scarcity of state-of-the-art Pt (and other noble metals) catalysts, pressure is growing to understand and improve the activities of the catalysts at minimum loadings and with maximum resistance to poisons and electrocatalytic interferences. Meier et al.8d demonstrated the complexity of degradation, on the nanoscale, of Vulcan carbon black supported Pt catalysts which leads to a loss of apparent electrochemically active surface area. In their work, four different degradation pathways were discussed: Pt agglomeration, carbon corrosion, Pt dissolution, and detachment of Pt particles (from the support). Liu and co-workers8c claimed that the adsorption and accumulation of the side-products (such as acetone) on electrodes' surfaces in an operating alkaline direct 2-propanol fuel cell were the reason for the poisoning of the Pt catalysts. Hidai et al. concluded that the dissolution of Pt hydroxide and oxide surface species and redeposition of Pt (in a reduced metallic state) were responsible for catalyst degradation.8a
In this initial study (of a planned series of studies), the effect of a selection of commonly encountered quaternary ammonium-, imidazolium-, and DABCO-based (AAEM-relevant) cationic groups on the oxygen reduction reaction (ORR) on a polycrystalline Pt (Ptpc) disk electrode has been investigated in strongly alkaline aqueous KOH (1 mol dm−3). The structures of the cationic model molecules studied are presented in Scheme 1. Techniques used include cyclic voltammetry (CV) and rotating-disk electrode (RDE) linear sweep voltammetry (LSV). We acknowledge that the electrochemical performances reported for these ex situ (aqueous electrolyte) studies will not directly translate into the catalytic performances in actual all-solid-state cells (where spectator ions, such as K+ and excess OH− ions, are not present): this is a subject discussed in detail by Kucernak et al.,12 who argue that all-solid-state electrochemical cells allow a more realistic ex situ measurement of electrocatalytic performances (that are more directly linked to in situ [fuel cell] performances). However, this paper produces useful initial data that indicate that AAEMs containing imidazolium-based (and additionally benzyl-based) functional group chemistries may not be suitable for use in alkaline polymer electrolyte fuel cells. This is especially the case when considering the growing evidence in the literature that imidazolium-type cationic groups may be significantly less stable in the presence of hydroxide anions than the most commonly encountered [and benchmark] quaternary ammonium groups.2a,d
![The structures of the model molecules bearing cationic head-groups: TMA = tetramethylammonium, BTMA = benzyltrimethylammonium, BMI = 1-benzyl-3-methylimidazolium, BAABCO = 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane, and BOTMHA = 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium. Cl− counter anions were used throughout.](/image/article/2013/CP/c3cp50556a/c3cp50556a-s1.gif) | ||
| Scheme 1 The structures of the model molecules bearing cationic head-groups: TMA = tetramethylammonium, BTMA = benzyltrimethylammonium, BMI = 1-benzyl-3-methylimidazolium, BAABCO = 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane, and BOTMHA = 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium. Cl− counter anions were used throughout. | ||
Future work will involve the extension of these initial observations to the study of fuel cell grade Pt/C and Pt black nanocatalysts (as opposed to a Ptpc disk), cationic groups located on insoluble (surface bound) ionomers used as catalyst binders (as opposed to small molecules dissolved in aqueous solution), electrolytes containing varying degrees of carbonate and bicarbonate alkaline anions, and the effect on the HOR as well as the ORR.
Experimental
Chemicals and materials
Aqueous KOH solution (1 mol dm−3), KCl, benzyltrimethylammonium chloride (BTMA), 1-benzyl-3-methylimidazolium chloride (BMI), and tetramethylammonium chloride (TMA) were obtained from Sigma-Aldrich and used as received. Both 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium (BOTMHA) and 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane (BAABCO) were in-house synthesised (see ESI†). All aqueous electrolyte solutions used in electrochemical experiments contained aqueous KOH (1 mol dm−3) with and without the addition of the cationic molecules (1 mmol dm−3) under study with control experiments involving aqueous KOH (1 mol dm−3) containing KCl (1 mmol dm−3). N2 and O2 purging of the electrolytes was conducted where appropriate and as detailed below. 18.2 MΩ cm water was used for electrode cleaning as well as for all rinsing steps.Electrochemical instrumentation
Voltammetric measurements were performed in a conventional three-electrode water-jacketed electrochemical cell (Princeton Applied Research, supplied by Ametek). Reference electrodes were HydroFlex Reversible Hydrogen Electrodes (RHE) obtained from Gaskatel (Germany). The performance (potential) of the RHE was continuously checked (before and after each experiment) using a saturated calomel laboratory master reference electrode (Gamry Instruments, USA). An annealed Pt wire (Advent Research Materials) with diameter ϕ = 0.5 mm and length = 0.5 m was coiled and used as an auxiliary electrode. The working electrode was a commercial Ptpc rotating disk electrode (RDE) with a geometrical surface area = 0.196 cm2 (Princeton Applied Research, supplied by Ametek). Obviously, due to the inherent nature of polycrystalline Pt, not every PtPt RDE will be exactly the same! Thus, for this study of the relative effects of addition of the different species, all of the data in this study was collected on the same RDE.Prior to each experiment, the Ptpc RDE was gently polished with 0.05 μm alumina slurry (BASi) to a mirror-like surface, and rinsed intensively with 18.2 MΩ cm water. The Ptpc electrode was then electrochemically pre-treated by CV cycling in N2-saturated aqueous KOH (1 mol dm−3) over a potential range of 0.05–1.2 V vs. RHE for 20 cycles at a sweep rate of 50 mV s−1 or until a stable CV response was obtained. Before each experiment, the repeatability of the Ptpc electrode response was strictly ensured by repeating the above electrochemical cleaning procedure and monitoring the final stable CV responses in aqueous KOH (1 mol dm−3).
The potentials were controlled using an Iviumstat potentiostat (Ivium Technologies Netherlands, supplied by Alvatek, UK) using IviumSoft 2.031 control software. The rotation speed of the RDE working electrode was varied from 400–2000 rpm using a Ring-Disk Electrode System model 636 rotator (Princeton Applied Research, supplied via Ametek). All experiments were conducted at a controlled temperature of 25 °C. Measurements were iR-corrected for the uncompensated ohmic drops by means of recording electrochemical impedance spectra from 100 → 0.1 kHz and with a voltage perturbation of 10 mVr.m.s.. The high frequency intercept of the real part of the Nyquist plot was taken as the uncompensated resistance.
Cyclic voltammetry
The aqueous electrolytes being used were deaerated by thorough purging with N2 gas and ensuring the N2 atmosphere above the electrolytes was maintained throughout the experiments. CV curves were recorded by cycling in the potential region 0.05–1.2 V vs. RHE at a scan rate of 50 mV s−1; wider potential windows were also probed by increasing the anodic (upper) potential in steps of 0.05 V. The Electrochemical Active Surface Area (ECSA cm−2) of the Ptpc disk electrode was determined from the integrated charge required for hydrogen adsorption–desorption in the CVs cycled in the potential region of 0.05–1.2 V vs. RHE (eqn (1)): | (1) |
The roughness factors were measured from CVs recorded using N2-saturated aqueous KOH (1 mol dm−3) solutions immediately before the relevant RDE experiments (i.e. directly before the addition of any of the organic cationic molecules or KCl). The roughness factor Rf of the Ptpc was calculated using eqn (2) with the data from the KOH (1 mol dm−3) measurements without additional species present:
 | (2) |
 | ||
| Fig. 1 The roughness factor for the polycrystalline Pt (Ptpc) disk electrode in N2-saturated aqueous KOH (1 mol dm−3) electrolytes at 25 °C before addition of 1 mmol dm−3 of the bracketed model head-group species (or KCl). Scan rate = 50 mV s−1. Error bars are the confidence intervals at the 95% confidence level (n = 3 repeated experiments). | ||
The ECSA measured in aqueous KOH (1 mol dm−3), before addition of organic cationic molecules or KCl, was used to normalise the current densities in the ORR RDE linear sweep voltammetry experiments detailed below. The apparent ECSAs were also calculated from the CVs with N2-purged electrolytes collected with the addition of the organic cationic species or KCl: this was to measure the percentage apparent ECSA remaining after addition of these additives (with the assumption that the hydrogen desorption under such conditions [in the presence of such species] is a reliable measure of ECSA).
Oxygen reduction reaction (ORR) hydrodynamic rotating disk electrode (RDE) linear sweep voltammetry (LSV)
The aqueous KOH electrolytes (with and without the addition of cationic species [and KCl] in concentrations of 1 mmol dm−3) were initially deaerated by thorough purging with N2 gas followed by a thorough O2 purge in order to obtain O2-saturation. An O2 atmosphere was maintained above the electrolyte surface throughout the subsequent measurements (gentle flow of O2 that did not disturb the electrolyte surface). The ORR activities of the Ptpc RDE were evaluated using LSV: the potentials were swept from 1.00 V to 0.2 V vs. RHE at a scan rate of 5 mV s−1; the experiments were repeated with rotation rates increasing from 400 to 2000 rpm in 200 rpm steps. Kinetic currents were determined using the Koutecký–Levich equation (eqn (3))13 for processes under mixed activation and mass-transport control: | (3) |
E = E0 − b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logik logik | (4) |
| E0 = Er + blogi0 | (5) |
| B = 0.2nAGEOFCoDo2/3ν−1/6 | (6) |
485 C mol−1), Co is the bulk concentration of O2 dissolved in the electrolyte (taken here as = 8.60 × 10−7 mol cm−3), Do is the O2 diffusion coefficient (taken here as = 2.04 × 10−5 cm2 s−1), and ν is the kinematic viscosity (taken here as = 0.93 cm2 s−1); these values were taken from the literature.15 The main assumption used for this simplistic analysis is that the added species were not affecting the properties of bulk KOH (1 mol dm−3).Results and discussion
Cyclic voltammetry
The effect of 1 mmol dm−3 concentrations of KCl, TMA, BMI, BTMA, BAABCO, and BOTMHA in the aqueous KOH (1 mol dm−3) electrolyte on the electrochemical behaviour of the bare Ptpc disk electrode was initially studied using cyclic voltammetry: the results are presented in Fig. 2 and the raw CV data (n = 3 replicates) can be found in Fig. ESI1 (ESI†). The changes in the CVs in the N2-purged electrolytes for the following potential regions are discussed below: (1) Pt-hydrogen adsorption–desorption region (0.05–0.45 V vs. RHE); (2) double-layer region (0.45–0.6 V vs. RHE); and (3) Pt-oxide region (>0.6 V vs. RHE). | ||
| Fig. 2 Cyclic voltammograms of a Ptpc disk electrode in N2-saturated aqueous KOH (1 mol dm−3) electrolytes at 25 °C with and without addition of 1 mmol dm−3 of cationic molecules and KCl (control experiment for Cl− spectator anions). Scan rate = 50 mV s−1. | ||
The Pt-hydrogen peaks at 0.27 V vs. RHE and 0.38 V vs. RHE with the KCl and TMA containing electrolytes were similar to those for the blank KOH (no additives); however, they were suppressed with BOTMHA, poorly defined with the presence of BTMA and BAABCO, and totally suppressed with the electrolyte containing BMI. The Pt-hydrogen adsorption–desorption regions in the CVs of BMI-, BTMA-, and BAABCO-containing electrolytes (i.e. with additives where the N atom was bound directly to the benzyl CH2 group) were significantly different in form compared to the other electrolytes and exhibited a characteristic flatter (plateau) topography. Additionally, the onset potential for the Pt-oxide reduction wave shifted to less positive potentials in the media containing the larger (benzene-ring-containing) cationic species. The anodic currents increased at potentials >+1.0 V vs. RHE [above the levels for Pt-oxidation as measured in additive-free aqueous KOH (1 mol dm−3) solutions] in the CVs of the BMI- and BOTMHA-containing electrolytes, with even higher currents with the BTMA- and BAABCO-containing electrolytes. However, there was no positive correlation between the intensity of these oxidation peaks (0.6−1.2 V vs. RHE) and the Pt-oxide-derived cathodic peak intensities (with peak potentials [Ep] of ca. 0.8 V vs. RHE) observed in the reverse scan direction.
However, if the CV potentials on the forward sweep were not allowed to reach the Pt-oxide formation plateau region (i.e. Pt surface oxidation has initiated but not plateaued), the intensities of the oxide reduction peaks were smaller with the presence of BMI, BTMA, BAABCO and BOTMHA; this indicates that the surface coverage of Pt-oxide species in the presence of BMI, BTMA, BAABCO, and BOTMHA was reduced compared to when additive-free KOH was used and when (benzene ring free) KCl and TMA species were present.
Fig. 3 compares the effect of 1 mmol dm−3 concentrations of BMI, BTMA, BAABCO and BOTMHA in the aqueous KOH (1 mol dm−3) electrolyte on the CV behaviour of the Ptpc disk electrode cycled at progressively higher anodic potentials. The anodic currents at the highest potentials (forward scans) and the cathodic currents in the double layer region and for hydrogen adsorption (in the reverse scans) all increased in magnitude upon increasing the upper anodic potential limits (Ef). In addition, the reduction peaks in the reverse scans, in the potential range 0.9–0.55 V vs. RHE, all increased in intensity and shifted to less positive peak potentials when the upper anodic potential limits were increased. The shifts were especially significant when the anodic upper potential limit was higher than 1.20 V vs. RHE. The anodic peaks reached a maximum current at ca. 1.3 V vs. RHE for the BTMA- and BAABCO-containing electrolytes, but the maximum anodic peak current was not observed until 1.40 V vs. RHE for the BOTMHA-containing electrolyte; however, no real plateau was observed with the BMI-containing electrolyte even at 1.5 V vs. RHE. These data are highly indicative of the oxidation of the organic cationic species containing benzene rings.
 | ||
| Fig. 3 Cyclic voltammograms of a polycrystalline Pt disk electrode in the N2-saturated aqueous KOH (1 mol dm−3) electrolyte at 25 °C without (--- dashed lines) and with addition of 1 mmol dm−3 of (A) BMI, (B) BTMA, (C) BAABCO, and (D) BOTMHA (— solid lines). The CVs with these 4 additives were recorded with progressively higher upper potential limits (0.05 V step size). Scan rate = 50 mV s−1. | ||
Charge densities QR were calculated by integration of the reduction peaks (in the potential range 0.9–0.55 V vs. RHE) in the reverese scans of CVs in Fig. 3 [with normalisation to the ECSAs of the Ptpc electrode when measured in additive-free aqueous KOH (1 mol dm−3) prior to addition of the cationic species (and KCl)]: Fig. 4 presents these data as a function of Ef. Inflection points in the plot were observed in the Ef 1.25–1.35 vs. RHE (reduction charge density range 61–212 μC cm−2). The degree of the inflection (gradient change) increased in the order: KOH < BOTMHA < BTMA < BAABCO < BMI. The presence of these inflection points indicates that more than one oxidation process occurs on the forward sweep and that the second process becomes dominant after certain anodic potentials.16 It is speculated that the two process would be Pt-oxide surface formation and oxidation of the organic cationic molecules. The data suggest that BMI inhibits surface Pt-oxide formation and that BMI oxidation initiates at higher anodic potentials (compared to the BTMA, BAABCO, and BOTMHA); this accounts for the especially large inflection (increase in the gradient) seen in Fig. 4 for BMI.
 | ||
| Fig. 4 Charge densities QR obtained by integration of the reduction peaks (in the potential range 0.9–0.55 V vs. RHE) in the reverese scans of CVs (in Fig. 3) with the Ptpc disk electrode recorded at 25 °C in N2-purged 1 mol dm−3 KOH (×) and with the addition of 1 mmol dm−3 of: (●) BMI, (◆) BTMA, (+) BAABCO and (△) BOTMHA at increasing upper anodic potential limits (Ef). | ||
The oxidation of the organic cationic molecules represents a complication to the experimental rationale of this study: this is because oxidised species will also be present (along with the intentionally introduced cationic species) and will represent an additional electrochemical interference in the ORR RDE LSV studies. These data suggest that, for the ORR RDE LSV studies, the electrodes need to be poised at as low as potential that is possible (for the shortest amount of time) before the ORR reduction (high to low potential) LSV sweeps are recorded.
Fig. 5 presents the apparent percentage ECSA remaining (% ECSARemain) when the PtPc electrode is studied in the electrolytes containing the additives when normalised to the initial ECSAs measured in the additive-free blank KOH electrolyte. The assumption made here is that the suppression of the H2 adsorption–desorption regions is due to the additives causing a lowering of the ECSA. The CV-calculated losses of ECSA with PtPc in the BMI- and BAABCO-containing electrolytes were higher than the already significant ECSA losses observed for BTMA- and BOTMHA-containing electrolytes. These results suggest that the benzene-ring-containing cationic molecules (BMI, BTMA, and BAABCO, and BOTMHA) significantly interfere with the electrochemical activity of the Ptpc surface when present in 1 mmol dm−3 concentrations in strongly alkaline aqueous KOH (1 mol dm−3). The adsorption of the head-groups hinders the adsorption–desorption of hydrogen on the Ptpc surface and reduces the apparent ECSAs. This electrochemical interference phenomena increase from TMA (zero measurable effect) < BOTMHA < BTMA < BAABCO for the quaternary ammonium species; the presence of a benzene ring appears to be significant and the linking of the quaternary ammonium to the benzene ring via a short –CH2– bridge is clearly detrimental: a longer hexyloxy spacer chain between the benzene ring and the quaternary ammonium group provided a small mitigation to this negative interference effect.
 | ||
| Fig. 5 The percentage ECSA remaining (% ECSARemain) at 25 °C of a polycrystalline Pt disk electrode calculated by comparing the ECSAs derived from the data in Fig. 2 (from the hydrogen desorption peaks in the CVs with the aqueous KOH (1 mol dm−3) electrolytes containing additives) with the ECSAs derived from (immediately) prior measured CVs using additive-free aqueous KOH (1 mol dm−3). Error bars are the confidence intervals at the 95% confidence level (n = 3 repeated experiments). | ||
The worst case scenario in this study is with the presence of an imidazolium ring (the BMI-containing electrolyte) where nearly all of the Ptpc surface area appeared to be electrochemically inactive (towards hydrogen adsorption and desorption). This initial finding suggests that imidazolium-based head-groups have a stronger negative effect on Ptpc in high pH aqueous electrolytes compared to quaternary ammonium-type head-groups. Interestingly, the presence of KCl (at 1 mmol dm−3 concentration) did not lead to measurable Ptpc ECSA inhibition in the strongly alkaline aqueous electrolytes: this is an interesting result as it is widely acknowledged17 that Cl− anions interfere with Pt electrocatalysts in acidic media.
Oxygen reduction reaction hydrodynamic voltammetry
Fig. 6 present the ECSA-normalised ORR RDE LSV at 1600 rpm: the ECSAs were measured from the blank N-purged aqueous KOH (1 mol dm−3) CVs recorded immediately before the addition of each cationic (and KCl) species and prior to O2-purging. The raw data at all rotation rates can be found in the ESI† as Fig. ESI2. The ORR is under mixed [kinetic and diffusion] control in the potential range 0.85–0.7 V vs. RHE and is kinetically controlled for potentials >0.9 V vs. RHE. Similar onset potentials of ca. 0.95 V vs. RHE were observed for all of the electrolytes studied. Since the specific surface areas of the Ptpc in the RDE for each experiment did not vary significantly (consistent roughness factors as shown in Fig. 1), the currents obtained were assumed to relate only to the nature of the electrolytes used.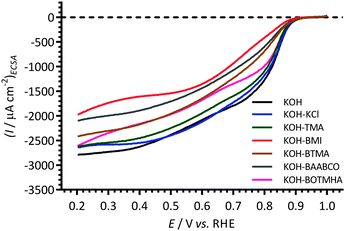 | ||
| Fig. 6 The ECSA-normalized linear sweep hydrodynamic voltammograms at 25 °C of the Ptpc disk RDE in the O2-saturated aqueous KOH (1 mol dm−3) electrolytes with and without addition of 1 mmol dm−3 of each of the cationic molecules studied and KCl (Cl− control experiment). Scan rate = 5 mV s−1 and the RDE rotation rate = 1600 rpm. The ECSAs were measured from the blank N-purged aqueous KOH (1 mol dm−3) CVs recorded immediately before the addition of each cationic (and KCl) species and prior to O2-purging. | ||
A relatively consistent ORR activity was observed for the blank KOH and with the KCl- and TMA-containing electrolytes. BTMA, BAABCO, and (especially) BMI significantly inhibit the ORR reaction on Ptpc. The Ptpc RDE showed the lowest ORR activity with the BMI-containing electrolyte with both geometric and ECSA normalised data (geometric normalised data not shown). The results show a reasonable correlation with the degree of inflection observed in Fig. 4 and the % ECSARemain presented in Fig. 5. When the current densities were normalised to the geometric surface areas (roughness factor not taken into account – data not shown), the ORR activity of Ptpc in the electrolytes containing BTMA and BAABCO appears to be identical.
The ORR electrocatalytic behaviour of the Ptpc RDE in KOH aqueous electrolytes with and without the different additives was further analysed using Tafel plots (Fig. 7) derived from the ORR RDE LSVs. The kinetic current density (ik) values were calculated using Koutecky–Levich analysis (eqn (3): raw data presented in the ESI† as Fig. ESI3). The ik were normalised to the ECSAs (as with the data in Fig. 6). As anticipated from the data presented in Fig. 6, the ORR activities of the Ptpc disk electrode exhibited clear negative horizontal shifts in the following sequence: blank KOH ≈ KOH-KCl < KOH-TMA ≈ KOH-BOTMHA < KOH-BTMA < KOH-BAABCO < KOH-BMI. Two quasi-linear Tafel regions with different Tafel slopes were observed. In the low overpotential region (>0.8 V vs. RHE), the Tafel slopes were close to −60 mV dec−1; the Tafel slopes in the higher overpotential region (in the range 0.8–0.75 V vs. RHE) were higher at ca. −120 mV dec−1. The precise Tafel slopes in these potential regions are summarised in Table 1.
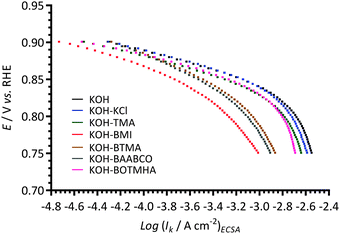 | ||
| Fig. 7 The Tafel plots at 25 °C of the Ptpc disk RDE in the O2-saturated aqueous KOH (1 mol dm−3) electrolytes with and without addition of 1 mmol dm−3 of each of the additives (derived from the data in Fig. ESI2, ESI†). The ECSAs were measured as described in the caption to Fig. 6. | ||
| O2-saturated aqueous electrolyte | Low overpotential Tafel slopesa [mV dec−1] | High overpotential Tafel slopesa [mV dec−1] | Apparent i0b [mA cm−2] | nc |
|---|---|---|---|---|
| a Extracted from the Tafel plots based on ECSA-normalised kinetic current densities normalised to ECSAs that were measured from the blank N2-purged aqueous KOH (1 mol dm−3) CVs recorded immediately before the addition of each cationic (and KCl) species and prior to O2-purging.b Apparent exchange current densities were normalised as with the kinetic current densities.c Calculated using eqn (6). | ||||
| Blank KOH | −60 | −121 | 0.144 | 4.1 |
| KOH-KCl | −64 | −132 | 0.156 | 4.1 |
| KOH-TMA | −55 | −106 | 0.125 | 4.1 |
| KOH-BMI | −75 | −109 | 0.052 | 2.4 |
| KOH-BTMA | −82 | −121 | 0.055 | 3.7 |
| KOH-BAABCO | −84 | −123 | 0.044 | 3.7 |
| KOH-BOTMHA | −60 | −121 | 0.154 | 4.1 |
Table 1 collates the RDE-derived ORR electrochemical parameters. The low overpotential Tafel slopes (>0.8 V vs. RHE) increased in the order: KOH-TMA < KOH-blank ≈ KOH-BOTMHA < KOH-KCl < KOH-BMI < KOH-BTMA < KOH-BAABCO. However in the high overpotential region (0.8–0.75 V vs. RHE), the Tafel slopes increased in a slightly different order: KOH-TMA < KOH-BMI < KOH-blank ≈ KOH-BOTMHA ≈ KOH-BTMA ≈ KOH-BAABCO < KOH-KCl. The Tafel slopes for the TMA-containing electrolyte were always the lowest (in both regions). The apparent exchange current densities (i0) were calculated from the hydrodynamic RDE voltammetric data collected by solving the eqn (4) and (5); values were higher (>0.1 mA cmECSA−2) for the blank KOH benchmark and the KCl-, TMA-, and BOTMHA-containing electrolytes. The values decreased dramatically (<0.06 mA cmECSA−2) with the remaining electrolytes in the order: KOH-BTMA ≈ KOH-BMI > KOH-BAABCO. This result again confirms that the presence of BMI, BTMA and BAABCO (or their electrochemical oxidation products) represents a significant catalytic interference (apparent exchange current densities that <50% of the value observed with the additive-free blank KOH electrolyte). As well as the consistently poor activity performance observed when BMI was added, a decrease in the number of ORR electrons transferred was observed (n = 2.4): this suggests that the presence of such imidazolium species may lead to an enhancement in the amount of undesirable peroxide species being produced in the ORR.
The high overpotential Tafel slope region (with higher magnitude Tafel slopes) corresponds to Ptpc surfaces with low oxide coverage (vice versa for the low overpotential Tafel slope region); hence, oxide-coverage-related activation barriers for ORR are anticipated.17 The existence of a high coverage of adsorbed O and/or OH related species would be expected to lower the Tafel slopes and this appears to be the case with the TMA-containing electrolytes (55 and 106 mV dec−1 for the low and high overpotential Tafel slopes respectively). It is hypothesised that the reaction steps after the initial electron transfer step related to O2 adsorption (forming adsorbed O and/or OH related species) are rate-determining18 for Ptpc in the TMA-containing electrolyte. The relatively stronger adsorption of oxides in the TMA-containing electrolyte leads to comparable ORR kinetics compared to the blank KOH electrolyte and the control electrolyte containing KCl (1 mmol dm−3).
However for the case of BMI with higher oxide-coverage at high overpotentials (interpreted from the relatively low Tafel slope value of 109 mV dec−1), the retention of OH-related species on the catalyst leads to the risk of an undesirable increase in the formation of peroxide species via the 2-electron ORR pathway. The generation of excessive peroxide species will lead to the degradation of fuel cell components (polymer electrolytes) and therefore shorter cell lifetimes. Thus, the ability to successfully utilise imidazolium head-groups (modelled here using BMI) in the AAEMs and alkaline ionomer APEFCs is still to be proven.
Nonetheless, the poor ORR activity on Ptpc in BTMA- and BAABCO-containing aqueous KOH electrolytes (suggested by their generally higher Tafel slopes in both overpotential regions) suggests a different mechanism for O2 adsorption is operating along with the “blocking” of the electrode (lower % ECSARemain). The presence of a benzene-ring connected to the nitrogen atoms via a –CH2– link (modelled here by BTMA, BAABCO, and BMI) clearly depresses the electrocatalytic behaviour of Ptpc in aqueous KOH (1 mol dm−3) by affecting both the hydrogen adsorption–desorption (Fig. 2) and oxide formation (Fig. 4) regions. Surprisingly, the presence of a longer aliphatic spacer between the quaternary ammonium cationic head-group and the benzyl-group (modelled by BOTMHA) delivered a reduced negative performance impact. The impact of the benzene-ring-free TMA was negligible. These results strongly suggest that future APEFC research should focus on developing alkaline anion-exchange polymer electrolytes with aliphatic-based cationic head-groups (not imidazolium) as long as this does not lead to a concomitant decrease in the chemical (alkaline) stabilities of the head-groups.
Conclusions
The effect on a polycrystalline Pt (Ptpc) disk electrode in aqueous electrolytes based on KOH (1 mol dm−3) of 1 mmol dm−3 concentrations of various anion-exchange cationic functionalities [tetramethylammonium (TMA), benzyltrimethylammonium (BTMA), 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium (BOTMHA), 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane (BAABCO), and 1-benzyl-3-methylimidazolium (BMI)] was investigated; KCl was also tested as all of the above cationic organic molecules were in the Cl− anion forms. The level of suppression of the hydrogen adsorption–desorption features in the cyclic voltammograms followed the order: BMI > BAABCO > BTMA > BOTMHA > TMA, KCl, and additive-free KOH. The hindrance towards the oxygen reduction reaction specific electrocatalytic activities on Ptpc increases in the order: blank KOH < KOH-KCl < KOH-TMA < KOH-BOTMHA < KOH-BTMA < KOH-BAABCO < KOH-BMI (when roughness factors were taken into consideration).The presence of BMI was particularly severe and appeared to primarily alter the reaction pathway. The addition of the additives containing benzene rings led to an apparent loss in electrochemical active surface areas; separating the benzene ring from the N atoms in the cationic functional group provided some mitigation towards performance losses. Removing the benzene ring completely (TMA experiments) led to almost insignificant losses in performances.
These results demonstrate for the first time that very small concentrations (1 mmol dm−3) of species such as BTMA, BAABCO and especially BMI (or potentially their electrochemical oxidation products) can seriously inhibit the specific catalytic activities of polycrystalline Pt electrodes at high pH in aqueous electrolytes. Future investigations will consider fuel cell grade nanocatalysts, cationic groups located on insoluble (surface bound) ionomers as catalyst binders, electrolytes containing varying degrees of carbonate and bicarbonate alkaline anions, and the effect on the hydrogen oxidation reaction.
Acknowledgements
The authors thank the UK's Engineering & Physical Sciences Research Council for funding (Prof. Varcoe's EPSRC Leadership Fellowship Grant EP/1004882/1). The University of Surrey also provided funds as part of Michael Fox's final year undergraduate project (synthesis of 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane and 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium). Francesca Robertson and Anuska Mann are thanked for HRMS and Qinmin Zhang is thanked for NMR spectroscopy.Notes and references
- J. R. Varcoe, R. C. T. Slade, E. L. H. Yee, S. D. Poynton, D. J. Driscoll and D. C. Apperley, Chem. Mater., 2007, 19, 2686 CrossRef CAS.
- (a) O. I. Deavin, S. Murphy, A. L. Ong, S. D. Poynton, R. Zeng, H. Herman and J. R. Varcoe, Energy Environ. Sci., 2012, 5, 8584 RSC; (b) G. Couture, A. Alaarddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521 CrossRef CAS PubMed; (c) G. Merle, M. Wessling and K. Nikmeijer, J. Membr. Sci., 2011, 377, 1 CrossRef CAS PubMed; (d) O. M. M. Page, S. D. Poynton, S. Murphy, A. Ong, D. M. Hillman, C. A. Hancock, M. G. Hale, D. C. Apperley and J. R. Varcoe, RSC Adv., 2013, 3, 579 RSC.
- (a) O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753 CrossRef CAS PubMed; (b) J. Ran, L. Wu, X. Lin, L. Jiang and T. Xu, RSC Adv., 2012, 2, 4250 RSC; (c) Y. Zhang, J. Fang, Y. Wu, H. Xu, X. Chi, W. Li, Y. Yang, G. Yan and Y. Zhuang, J. Colloid Interface Sci., 2012, 381, 59 CrossRef CAS PubMed; (d) C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438 CrossRef CAS PubMed.
- (a) H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem., 2012, 116, 9419 CrossRef CAS PubMed; (b) J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399–400, 49 CrossRef CAS PubMed.
- (a) B. Qiu, B. Lin, L. Qiu and F. Yan, J. Mater. Chem., 2012, 22, 1040 RSC; (b) M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013 DOI:10.1002/polb.23149; (c) N. Li, Q. Zhang, C. Wang, Y. M. Lee and M. D. Guiver, Macromolecules, 2012, 45, 2411 CrossRef CAS; (d) B. S. Ko, J. Y. Sohn and J. Shin, Polymer, 2012, 53, 4652 CrossRef CAS PubMed; (e) C. G. Arges, J. Parrondo, G. Johnson, A. Nadhan and V. Ramani, J. Mater. Chem., 2012, 22, 3733 RSC; (f) B. Lin, L. Qiu, B. Qiu, Y. Peng and F. Yan, Macromolecules, 2011, 44, 9642 CrossRef CAS; (g) X. Lin, L. Wu, Y. Liu, A. L. Ong, S. D. Poynton and J. R. Varcoe, J. Power Sources, 2012, 217, 373 CrossRef CAS PubMed; (h) M. Guo, J. Fang, H. Xu, W. Li, X. Lu, C. Lan and K. Li, J. Membr. Sci., 2010, 362, 97 CrossRef CAS PubMed; (i) J. Ran, L. Wu, J. R. Varcoe, A. L. Ong, S. D. Poynton and T. Xu, J. Membr. Sci., 2012, 415–416, 242 CrossRef CAS PubMed; (j) X. Yan, G. He, S. Gu, X. Wu, L. Du and Y. Wang, Int. J. Hydrogen Energy, 2012, 37, 5216 CrossRef CAS PubMed; (k) J. Fang, Y. Yang, X. Lu, M. Ye, W. Li and Y. Zhang, Int. J. Hydrogen Energy, 2012, 37, 594 CrossRef CAS PubMed.
- A. Sarkar, X. Zhu, H. Nakanishi, J. B. Kerr and E. J. Cairns, J. Electrochem. Soc., 2012, 159, 628 CrossRef PubMed.
- (a) C. K. Poh, S. H. Lim, Z. Tian, L. Lai, Y. P. Feng, Z. Shen and J. Lin, Nano Energy, 2013, 2, 28 CrossRef CAS PubMed; (b) S. Cherevko, X. Xing and C. H. Chung, Electrochim. Acta, 2011, 56, 5771 CrossRef CAS PubMed; (c) A. Ilie, M. Simoes, S. Baranton, C. Coutanceau and S. Martemianov, J. Power Sources, 2011, 196, 4965 CrossRef CAS PubMed.
- (a) S. Hidai, M. Kobayashi, H. Niwa, Y. Harada, M. Oshima, Y. Nakamori and T. Aoki, J. Power Sources, 2012, 215, 233 CrossRef CAS PubMed; (b) C. Contanceau, P. Urchaga and S. Baranton, Electrochem. Commun., 2012, 22, 109 CrossRef PubMed; (c) Y. Liu, Y. Zeng, R. Liu, H. Wu, G. Wang and D. Cao, Electrochim. Acta, 2012, 76, 174 CrossRef CAS PubMed; (d) J. C. Meier, C. Galeano, I. Katsounaros, A. A. Topalov, A. Kostka, F. Schüth and K. J. J. Mayrhofer, ACS Catal., 2012, 2, 832 CrossRef CAS.
- (a) C. V. Rao and Y. Ishikawa, J. Phys. Chem. C, 2012, 116, 4340 CrossRef; (b) S. M. Alia, K. Duong, T. Liu, K. Jensen and Y. Yan, ChemSusChem, 2012, 5, 1619 CrossRef CAS PubMed; (c) J. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333 RSC; (d) J. Sunarso, A. A. J. Torriero, W. Zhou, P. C. Howlett and M. Forsyth, J. Phys. Chem. C, 2012, 116, 5827 CrossRef CAS.
- (a) Y. Zha, M. L. Disabb-Miller, Z. D. Johndon, M. A. Hickner and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 4493 CrossRef CAS PubMed; (b) N. Li, T. Yan, Z. Li, T. Thurn-Albrecht and W. H. Binder, Energy Environ. Sci., 2012, 5, 7888 RSC; (c) B. K. Balan and S. Kurungot, J. Mater. Chem., 2011, 21, 19039 RSC.
- (a) N. Follain, S. Roualdes, S. Marais, J. Frugier and M. Reinholdt, J. Phys. Chem. C, 2012, 116, 8510 CrossRef CAS; (b) G. K. S. Prakash, F. C. Kause, F. A. Viva, S. R. Narayanan and G. A. Olah, J. Power Sources, 2011, 196, 7967 CrossRef CAS PubMed.
- A. R. Kucernak and E. Toyoda, Electrochem. Commun., 2008, 10, 1728 CrossRef CAS PubMed; A. E. S. Sleighthome, J. R. Varcoe and A. R. Kucernak, Electrochem. Commun., 2008, 10, 151 CrossRef PubMed.
- C. H. Hamann, A. Hamnett and W. Vielstich, Electrochemistry, 2nd edn, Wiley-VCH, Weinheim, 2007, p. 1 Search PubMed.
- S. Srinivasan, E. A. Ticianelli, C. R. Derouin and A. Redondo, J. Power Sources, 1988, 22, 359 CrossRef CAS.
- J. A. Vega and W. E. Mustain, Electrochim. Acta, 2010, 55, 1638 CrossRef CAS PubMed.
- G. Macfie, A. Cooper and M. F. Cardosi, Electrochim. Acta, 2011, 56, 8394 CrossRef CAS PubMed.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654 RSC.
- A. C. Co, S. J. Xia and V. I. Birss, Oxygen reactions at Platinum/YTTRA-stabilized zirconia (YSZ) interfaces, The Electrochemical Society Inc., Pennington, New Jersey, 2001, p. 141 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: This includes a selection of relevant raw electrochemical data (CV and RDE voltammograms and K–L plots) as well as details on the synthesis of 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium (BOTMHA) and 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane (BAABCO). See DOI: 10.1039/c3cp50556a |
| This journal is © the Owner Societies 2013 |