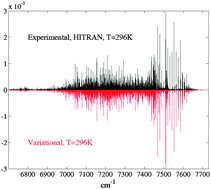
We report global calculations of rovibrational spectra and dipole transition intensities of methane using our recent ab initio dipole moment and potential surfaces [Nikitin et al., Chem. Phys. Lett., 2011, 501, 179; 2013, 565, 5]. For the full symmetry account, a recently published variational tensor formalism in normal modes [Rey et al., J. Chem. Phys., 2012, 136, 244106] is applied, the convergence of high-J calculations being improved by the use of vibrational eigenfunctions to make a compressed basis set for solving the rovibrational problem. Comparisons of theoretical predictions up to J = 25 for various complex polyads of methane involving strongly coupled vibration–rotation bands support the validity of this new approach. For the first time, positions and line intensities at 80 K and 296 K are shown to be in excellent agreement with raw experimental data, even for high energy ranges. The theoretical predictions also correctly describe the isotopic effects in line positions and intensities due to the CH4 → CD4 substitution which is considered as the test for the method. This work is a first step toward the theoretical interpretation of numerous methane bands which remain still unassigned and detailed line-by-line absorption/emission spectra analyses for atmospheric and planetological applications.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


