Impact of long-range van der Waals forces on chiral recognition in a Cinchona alkaloid chiral selector system†
Received
10th December 2012
, Accepted 25th February 2013
First published on 18th March 2013
Abstract
Singly-charged complexes of (8S,9R)-tert-butylcarbamoylquinine (tBuCQN), N-3,5-dinitrobenzoyl-(S,R)-leucine (DNB-S/R-leucine), and alkali metal counter ions (Li+, Na+, K+) were investigated by density-functional theory. It is shown that the cations prefer formation of an ionic pair with the carboxylate group of DNB-Leu over the formation of a cation–π interaction. The [tBuCQN·DNB-S/R-Leu·Na]+ complex is bound by a Coulombic attraction, a hydrogen bond, a π−π interaction and van der Waals forces. The tBuCQN chiral selector preferentially complexes with the DNB-S-Leu enantiomer, because the favourable stereochemistry allows the stabilization of the complex by at least one binding mode more compared to the complex containing the DNB-R-Leu molecule. Weakening of the binding modes is observed using the lithium counter ion compared to the sodium one. The weakening is more pronounced in [tBuCQN·DNB-R-Leu·Li]+ than in [tBuCQN·DNB-S-Leu·Li]+. The exact opposite effect is observed using the potassium counter ion. Hence, the lithium counter ion enhances the enantioselectivity of tBuCQN while the potassium counter ion reduces the enantioselectivity of tBuCQN.
1. Introduction
Today, chiral compounds are used in large scales and in many fields of chemistry.1,2 Along with this, the identification and separation of enantiomers are important processes. High-performance liquid chromatography (HPLC) can be considered as one of the most popular techniques for chiral separations and it is considered as a reliable tool for direct chiral separations if appropriate chiral stationary phases (CSPs) are available.3 Synthesis of new chiral compounds requires development of new CSPs that can determine the ee yield of separated enantiomers. However, the development of new CSPs is an expensive process. A new compound must be synthesized in a sufficient amount and bound to a solid support to create a chromatographic column. Therefore, it is desirable to find an inexpensive and efficient screening method that would identify promising candidates. Mass spectrometry (MS) is a technique that allows routine, fast, and cheap analysis of samples. This fact has encouraged the development of mass spectrometric tools for investigating chiral recognition in the last decade.4–13 Methods of computational chemistry are another suitable approach. They are widely used in drug discovery as a cheap screening method14–16 or in biochemistry in order to shed light on the functions of enzymes.17
Herein, we present a computational study, using density functional theory to investigate structure and stability of non-covalently bound complexes of (8S,9R)-tert-butylcarbamoylquinine (tBuCQN), N-3,5-dinitrobenzoyl-(S,R)-leucine (DNB-S/R-Leu), and alkali cations in the gas phase (Fig. 1a). The tBuCQN molecule belongs to a family of quinine (Cinchona alkaloid) derivatives that have been successfully used as CSPs (Fig. 1b) for the separation of enantiomers of chiral acids, particularly N-blocked amino acids.18–20 This system has also been extensively studied by HPLC and electrospray ionization-mass spectrometry (ESI-MS) methods.2,9,21 Both molecules are bound together via non-covalent binding modes (Fig. 1b). Formation of the alkali complexes was observed in ESI-MS experiments and preferential formation of a diastereoisomeric complex containing the (S)-optical isomer of DNB-Leu was shown to correlate well with HPLC results. The ESI-MS experiments further showed that enantioselectivity depends on the alkali metal involved in the complex.22 Thus, the best enantioselectivity was observed for the lithium complexes while efficiency of the enantioselectivity was lower for the sodium and potassium complexes. Therefore, special attention is paid to the understanding of the role and the placement of the alkali metal ion in the diastereomeric complexes to achieve different levels of enantioselectivity in this work.
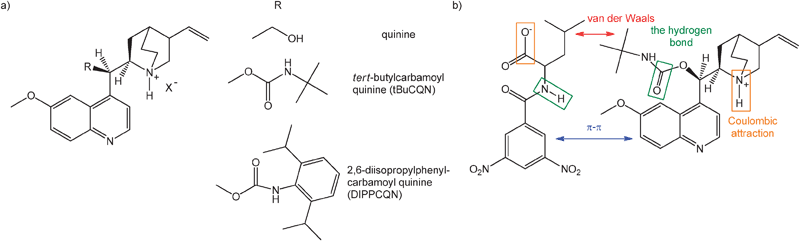 |
| | Fig. 1 (a) Representative Cinchona alkaloid quinine analogs, which have been investigated for use as chiral selectors. (b) Schematic representation of non-covalent binding interactions in the tBuCQN·DNB-S/R-Leu complex. The interacting groups are highlighted in the same colour. | |
2. Methods
All calculations were carried out using Gaussian 09 Revision A.02.23 The Becke-97D24 functional was used for geometry optimizations in conjunction with the 6-311++G** basis set.25–27 Density Fitting Basis Sets (DFBS) were generated using the automatic algorithm implemented in Gaussian 09 to reduce computational cost.28,29 The B97D functional is a pure GGA functional based on the Becke-97 (ref. 30) functional and includes the second version of Grimme's empirical dispersion correction. The empirical dispersion correction term is given by| | 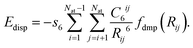 | (1) |
Here, Nat corresponds to the number of atoms in the system, C6ij is the dispersion coefficient for atom pair ij, s6 is a global scaling factor that depends only on the density functional used, Rij is the interatomic distance, and fdmp is a damping function that decays at small R fast enough to zero as the dispersion corrections are small between atoms below typical van der Waals distances and hence, “normal” bonds are not significantly affected by the correction. The fdmp function is defined by| | 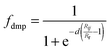 | (2) |
where Rr is the sum of atomic vdW radii and d is a parameter.
The B97D functional was successfully tested for variety of systems including the rare gas and benzene dimers, transition metal complexes and so forth (for more details see ref. 24 and 31). A benchmark study was published by Á. Vázquez-Mayagoitia et al.32 for benzene dimers and they showed that the B97D functional provides energies with errors less than 4.18 kJ mol−1, compared to CCSD(T). The B97D functional is thus a good compromise between the computational cost and the accuracy and was applied in different studies.33–35 Frequency calculations were done at the same level of theory to check the identity of all minima. Relative energies of the optimized structures were obtained by single-point energy calculations using the PBE0 functional (also denoted PBE1PBE) in conjunction with a 6-311++G** basis set and the “ultrafine” integration grid (i.e., a pruned (99,590) grid). The PBE0 functional is a hybrid version of the PBE functional incorporating 25% Hartree–Fock exchange.36–38 The relative energies are corrected for the dispersion effect by the third version of the Grimme empirical model.31 Partial atomic charges were obtained from Charge Model 5 (CM5) introduced by A. V. Marenich et al.39–43 that yields class IV charges derived from Hirshfeld population analysis.41 The NCIPLOT program, which was developed by Contreras-García et al., was used in an attempt to identify and visualize non-covalent interactions.44 It analyzes the reduced density gradient s of the electron density ρ obtained by the single-point energy calculations at low densities that are related to the weakest interaction. Therefore, a density cutoff of ρ < 0.1 a.u. was chosen as it includes the region of interest (see Fig. 3).
3. Results
Formation of singly charged complexes [tBuCQN·DNB-S/R-Leu·Na]+ was observed in previous ESI-MS experiments.2 Molecules are reasoned to be bound via four non-covalent interactions (Fig. 1b): (i) a Coulombic attraction of carboxylic oxygen O−(3) (Scheme 1) of DNB-Leu and the protonated amine group N(5)–H(4)+ of tBuCQN, (ii) a hydrogen bond between carbonyl of the carbamoyl group O(6) of tBuCQN and H(7) of the amide group of DNB-Leu, (iii) the π−π interaction (π stacking) of dinitrobenzoyl and 6-methoxy-quinolyl rings, and (iv) van der Waals interactions between the alkyl chains. The presence of sodium in the complexes raises a question about its location. The sodium atom can be rationally positioned at three potential binding sites in the complexes. Both molecules of the complexes have aromatic moieties that offer two binding modes for the sodium atom by a cation–π interaction.45 The third possibility is an ionic bond between the sodium atom and the carboxylate group of DNB-Leu.
![Schematic representation of the [tBuCQN·DNB-S/R-Leu·Na]+ complex.](/image/article/2013/CP/c3cp44444a/c3cp44444a-s1.gif) |
| | Scheme 1 Schematic representation of the [tBuCQN·DNB-S/R-Leu·Na]+ complex. | |
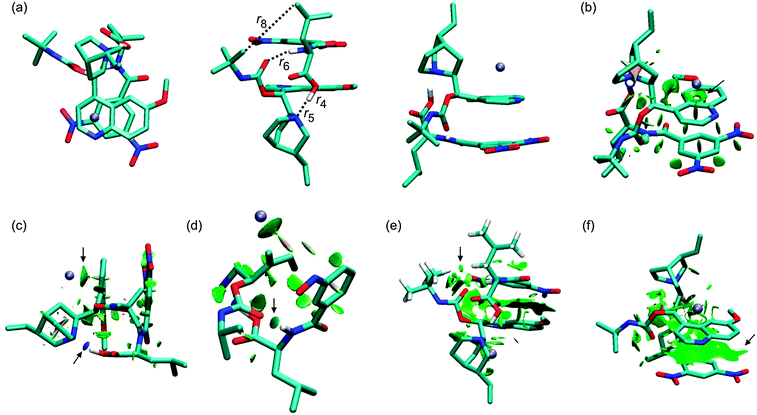
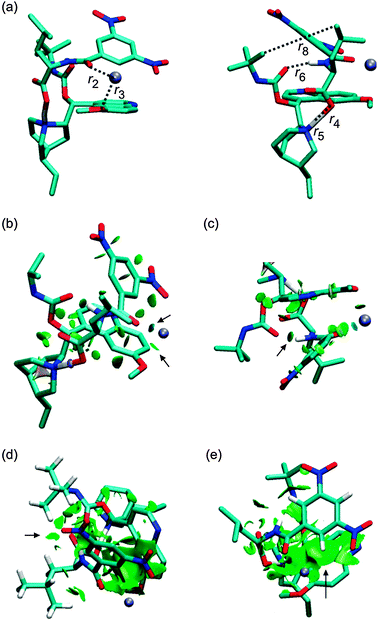
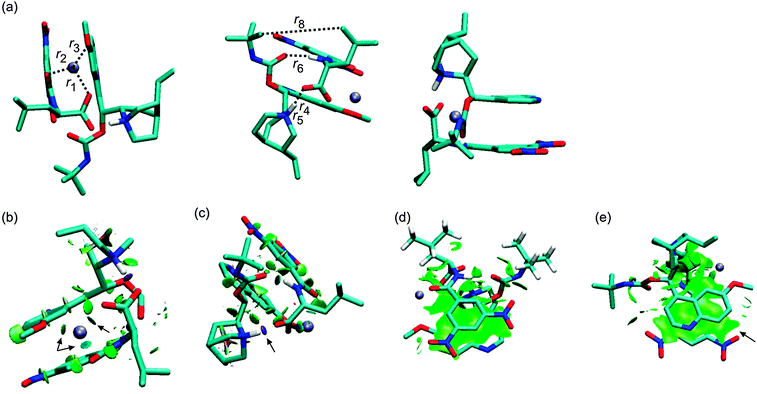
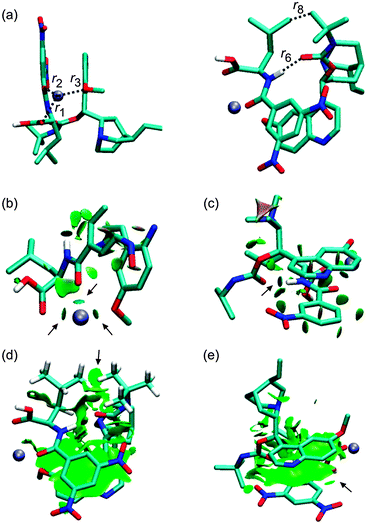
The crystal structure of a complex of β-chloro-tert-butylcarbamoylquinine and DNB-S-Leu was taken as a starting point for geometry optimizations.20 The original crystal structure was modified by replacing the chlorine atom with hydrogen and adding a sodium atom. It was found that a complex containing the Na+–−OOC pair (1SNa+; Fig. 2) is the most stable one (Table 1) compared to the other complexes with the cation–π interaction (2SNa+ and 3SNa+; Fig. 4 and 5). The sodium cation in 1SNa+ is coordinated in a pool of oxygen atoms (the amidic O(10), carbonyl O(1) of the DNB-S-Leu carboxylate group and the methoxy group O(11) of tBuCQN, Scheme 1), as shown by the three attractive isosurfaces surrounding the sodium cation (Fig. 2b). The next attractive isosurface belongs to the strong Coulombic interaction of the O−(3)–H+(4) pair. In the plot of the reduced density gradient versus the electron density multiplied by the sign of the second Hessian eigenvalue (so called NCI plot), a spike that corresponds to this interaction is significantly shifted to a negative value of the electron density with a peak at −0.074 a.u. (Fig. 3). The large shift indicates that the O−(3)–H+(4) pair is not a purely ionic bond and has some covalent character. An isosurface of an attractive interaction is found for the next binding mode, the hydrogen bond (Fig. 2c) in 1SNa+. The weak attractive interaction is revealed between the two alkyl chains (Fig. 2d) and another low density surface is between the overlapping portion of the ring moieties related to the π−π interaction (Fig. 2e). The π−π interaction is strong as indicated by the short ring–ring distance r9 (3.276 Å, Table 2). For comparison, the calculated distances for benzene- or benzene–pyridine sandwich dimers are in the order of 3.8–3.9 Å.46,47 Placing the sodium atom on the top of the 6-methoxy-quinolyl ring leads to a considerably less stable complex 2SNa+ (compared to 1SNa+). The sodium atom is positioned above the heterocyclic moiety and is bound by the cation–π interaction (Fig. 4b and c). The proton bound to the nitrogen atom of the bicyclic moiety and forming the O−(3)–H+(4) pair spontaneously migrates to the carboxylic group during the optimization (Fig. 4c). The 2SNa+ complex thus contains another hydrogen bond instead of the Coulombic attraction. Compared to the O(6)⋯H(7)–N(8) hydrogen bond, this N(5)⋯H(4)–O(3) hydrogen bond is very strong (Fig. 4c and d) with a spike at the reduced density gradient plot at −0.057 a.u. (see Fig. S1, ESI†). Furthermore, the charge distribution is influenced and the opposite direction of charge transfer is observed in that positive charge is on tBuCQN and DNB-S-Leu becomes negative (Table 3). Removing the sodium atom from the bonding distance of the carboxylic group of DNB-S-Leu has thus a significant impact on the entire complex. The O(6)⋯H(7)–N(8) hydrogen bond is weaker compared to 1SNa+ and the r8 distance is elongated that results only in a weak interaction of two hydrogen atoms (Fig. 4e) with respect to 1SNa+. Interestingly, no significant impact on the π−π interaction occurs (Fig. 4f). In the last tested isomer 3SNa+, the sodium cation is placed above the dinitrobenzoyl moiety. Nevertheless, the sodium cation is attracted by the oxygen atom O(10) of the amidic carbonyl during the optimization. In the final structure, it is coordinated to the oxygen atom O(10) of the amidic carbonyl via a strongly attractive interaction (Fig. 5b) and to the methoxy group and the π system of the quinolyl moiety (Fig. 5e). The O−(3)–H+(4) pair has more covalent character than in 1SNa+ (sign(λ2)ρ = −0.082; see Fig. S2, ESI†) and the O(6)⋯H(7)–N(8) hydrogen bond (Fig. 5c) is weaker than in 1SNa+. The alkyl groups are put apart compared to 1SNa+ with the large r8 distance and similar to 2SNa+ there is a weak interaction of two hydrogen atoms of the alkyl groups (Fig. 5d). The π−π interaction of the two aromatic rings is diminished (Fig. 5e). Hence hereafter, the most stable isomer 1SNa+ represents a reference for the following discussion of additional results.
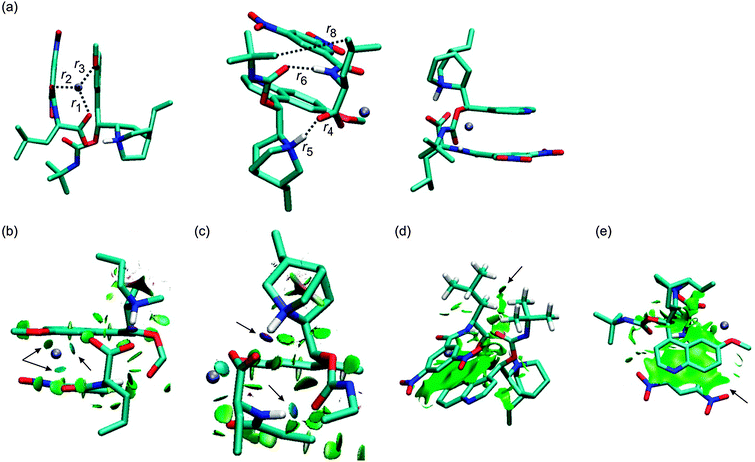 |
| | Fig. 2 The optimized structures of the 1SNa+ complex (B97D/6-311++G**/DFBS). Hydrogen atoms were removed except for the important ones to improve the clarity of the pictures. Colour codes: light blue, white, red, blue, and ice-blue correspond to carbon, hydrogen, oxygen, nitrogen, and sodium, respectively. Colour codes of the isosurfaces: the weak-, the strong attractive-, and the strong repulsive interactions are in green, blue, and red, respectively. The black arrows indicate the isosurfaces discussed in the text. | |
Table 1 Relative energies and entropies of the optimized complexes (obtained at the PBE0/6-311++G**//B97D/6-311++G**/DFBS level of theory)
| 1S/RX+ |
ΔrelG298 (kJ mol−1) |
ΔrelS298 (J mol−1 K−1) |
| 1SNa+ |
0.0 |
0.000 |
| 2SNa+ |
175.9 |
8.916 |
| 3SNa+ |
78.7 |
12.267 |
| 1RNa+ |
14.9 |
3.862 |
| 2RNa+ |
89.5 |
37.510 |
| 3RNa+ |
64.4 |
38.614 |
| 1SLi+ |
0.0 |
0.000 |
| 1RLi+ |
18.7 |
−12.221 |
| 1SK+ |
0.0 |
0.000 |
| 1RK+ |
6.5 |
2.753 |
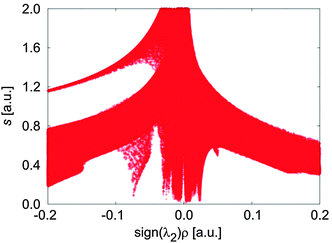 |
| | Fig. 3 Dependence of the reduced density gradient on the electron density multiplied by the sign of the second Hessian eigenvalue – so called NCI plot for 1SNa+. | |
Table 2 Monitored distances (Å) for the most stable structures of [tBuCQN·DNB-S/R-Leu·X]+ complexes, where X = Na, Li, and K
| 1S/RX+ |
r
1
|
r
2
|
r
3
|
r
4
|
r
5
|
r
4 + r5 |
r
6
|
r
7
|
r
6 + r7 |
r
8
|
r
9
|
| 1SNa+ |
2.247 |
2.279 |
2.416 |
1.531 |
1.093 |
2.624 |
1.839 |
1.023 |
2.862 |
4.257 |
3.276 |
| 2SNa+ |
|
|
|
1.026 |
1.698 |
2.724 |
1.926 |
1.018 |
2.944 |
5.190 |
3.282 |
| 3SNa+ |
|
2.243 |
2.515 |
1.498 |
1.114 |
2.612 |
1.940 |
1.024 |
2.964 |
5.848 |
— |
| 1RNa+ |
2.240 |
2.250 |
2.433 |
1.517 |
1.096 |
2.613 |
1.869 |
1.020 |
2.889 |
5.907 |
3.268 |
| 2RNa+ |
2.435 |
2.271 |
2.380 |
— |
— |
— |
1.845 |
1.027 |
2.872 |
3.612 |
3.301 |
| 3RNa+ |
2.424 |
2.403 |
2.388 |
1.501 |
1.000 |
2.501 |
— |
— |
— |
3.728 |
— |
| 1SLi+ |
1.885 |
1.911 |
2.088 |
1.539 |
1.088 |
2.627 |
1.859 |
1.021 |
2.880 |
4.428 |
3.267 |
| 1RLi+ |
1.869 |
1.898 |
2.093 |
1.543 |
1.087 |
2.630 |
1.909 |
1.019 |
2.928 |
5.952 |
3.270 |
| 1SK+ |
2.601 |
2.594 |
2.893 |
1.539 |
1.069 |
2.608 |
1.943 |
1.018 |
2.961 |
4.052 |
3.270 |
| 1RK+ |
2.745 |
2.697 |
2.861 |
1.499 |
1.091 |
2.590 |
1.838 |
1.017 |
2.855 |
4.432 |
3.289 |
Table 3 Selected group CM5 partial charges of complexes. X stands for Na, Li, and K
| 1S/RX+ |
X |
COO (DNB-Leu) |
CO (DNB-Leu) |
CTa (DNB-Leu) |
OCH3 (tBuCQN) |
The bicyclic residue (tBuCQN) |
CTa (tBuCQN) |
|
CT stands for charge transfer.
|
| 1SNa+ |
0.76 |
−0.61 |
−0.16 |
0.34 |
−0.03 |
0.69 |
−0.10 |
| 2SNa+ |
0.75 |
−0.13 |
−0.06 |
−0.10 |
0.02 |
0.20 |
0.35 |
| 3SNa+ |
0.69 |
−0.54 |
−0.14 |
0.42 |
0.01 |
0.65 |
−0.11 |
| 1RNa+ |
0.76 |
−0.64 |
−0.07 |
0.43 |
−0.09 |
0.68 |
−0.19 |
| 2RNa+ |
0.77 |
−0.05 |
−0.14 |
0.96 |
−0.07 |
0.08 |
−0.73 |
| 3RNa+ |
0.73 |
−0.64 |
−0.10 |
0.43 |
−0.03 |
0.67 |
−0.16 |
| 1SLi+ |
0.60 |
−0.58 |
−0.14 |
0.42 |
0.01 |
0.70 |
−0.02 |
| 1RLi+ |
0.76 |
−0.69 |
−0.08 |
0.41 |
−0.08 |
0.67 |
−0.17 |
| 1SK+ |
0.86 |
−0.64 |
−0.17 |
0.29 |
−0.08 |
0.71 |
−0.15 |
| 1RK+ |
0.84 |
−0.68 |
−0.14 |
0.28 |
−0.03 |
0.69 |
−0.11 |
 |
| | Fig. 4 The optimized structure of the 2SNa+ complex (B97D/6-311++G**/DFBS). Hydrogen atoms were removed except for the important ones to improve the clarity of the pictures. Colour codes: light blue, white, red, blue, and ice-blue correspond to carbon, hydrogen, oxygen, nitrogen, and sodium, respectively. Colour codes of the isosurfaces: the weak-, the strong attractive-, and the strong repulsive interactions are in green, blue, and red, respectively. The black arrows indicate the isosurfaces discussed in the text. | |
 |
| | Fig. 5 The optimized structure of the 3SNa+ complex (B97D/6-311++G**/DFBS). Hydrogen atoms were removed except for the important ones to improve the clarity of the pictures. Colour codes: light blue, white, red, blue, and ice-blue correspond to carbon, hydrogen, oxygen, nitrogen, and sodium, respectively. Colour codes of the isosurfaces: the weak-, the strong attractive-, and the strong repulsive interactions are in green, blue, and red, respectively. The black arrows indicate the isosurfaces discussed in the text. | |
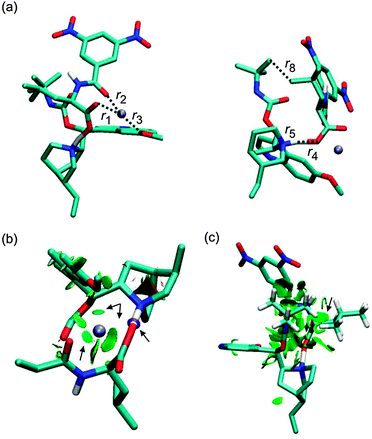
Replacing of the (S) optical isomer of DNB-Leu for the (R) isomer leads to complexes in which at least one binding mode is missing. It can be intuitively expected that switching of the hydrogen- and the alkyl positions at the chiral center of DNB-Leu will have an effect on the stability of [tBuCQN·DNB-Leu·Na]+. Indeed, the 1RNa+ complex (Fig. 6) is found to be energetically preferred over 2RNa+ and 3RNa+. The r1, r2 and r3 distances are shortened in 1RNa+ compared to 1SNa+. The strength of the interaction in the O−(3)–H+(4) pair is increased, which is reflected by a shortening of r4 and also by the increased value of sign(λ2)ρ, which is −0.077 a.u. (Fig. 6c, Fig. S3, ESI†). The stronger hydrogen bond is found in 1RNa+ than in 1SNa+ as shown by shorter r6 in 1RNa+ (Fig. 6c). In contrast, interaction of the two alkyl chains diminishes in 1RNa+ compared to 1SNa+ as indicated by the large value of r8 and also the missing isosurface of the weak interaction (Fig. 6d). The position of the dinitrobenzoyl ring is similar in 1RNa+ and 1SNa+ and there is only a small difference in r9 in these two complexes (Fig. 6e). The next considered complex contains the carboxylic group rotated away from the close distance of the protonated nitrogen. The rotation brings the alkyl groups closer together and results in a stronger van der Waals interaction. However, during the optimization, the Coulombic attraction prevailed over the vdW interaction and the optimization led again to the 1RNa+ isomer. The way to prevent this result is to neutralize the carboxylate group. Optimization of the suggested structure provided a stable isomer 2RNa+ (Fig. 7). The sodium atom remains coordinated by three oxygen atoms similarly to 1SNa+ (Fig. 7b) but the carboxylic group is neutral, which leads to a missing spike related to the O−(3)–H+(4) pair interaction in the NCI plot (Fig. S4, ESI†). The O(6)⋯H(7)–N(8) bond (Fig. 7c) is slightly affected as r6 is elongated. Interaction of the alkyl chains (Fig. 7d) increases with respect to 1SNa+ while the π−π interaction (Fig. 7e) is weaker than in 1SNa+. Therefore, the Coulombic interaction of the O−(3)–H+(4) pair has a significant impact on the overall stability of [tBuCQN·DNB-Leu·Na]+. At the last, an isomer, 3RNa+, with the broken O(6)⋯H(7)–N(8) hydrogen bond and the π−π interaction is considered (Fig. 8). Again there is a weakening of an interaction of the sodium cation with the oxygen atoms compared to the binding situation in 1SNa+ (Fig. 8b). The 3RNa+ isomer contains a stronger interaction in the O−(3)–H+(4) pair than in 1SNa+ (Fig. 8b and Fig. S5, ESI†) and also significantly shorter r8 (Fig. 8c). The missing hydrogen bond and the π−π interaction lead to an isomer with an energetic stability between 1RNa+ and 2RNa+. Clearly, both binding modes contribute to the stability of [tBuCQN·DNB-Leu·Na]+ less than the Coulomb interaction.
 |
| | Fig. 6 The optimized structure of the 1RNa+ complex (B97D/6-311++G**/DFBS). Hydrogen atoms were removed except for the important ones to improve the clarity of the pictures. Colour codes: light blue, white, red, blue, and ice-blue correspond to carbon, hydrogen, oxygen, nitrogen, and sodium, respectively. Colour codes of the isosurfaces: the weak-, the strong attractive-, and the strong repulsive interactions are in green, blue, and red, respectively. The black arrows indicate the isosurfaces discussed in the text. | |
 |
| | Fig. 7 The optimized structure of the 2RNa+ complex (B97D/6-311++G**/DFBS). Hydrogen atoms were removed except for the important ones to improve the clarity of the pictures. Colour codes: light blue, white, red, blue, and ice-blue correspond to carbon, hydrogen, oxygen, nitrogen, and sodium, respectively. Colour codes of the isosurfaces: the weak-, the strong attractive-, and the strong repulsive interactions are in green, blue, and red, respectively. The black arrows indicate the isosurfaces discussed in the text. | |
 |
| | Fig. 8 The optimized structure of the 3RNa+ complex (B97D/6-311++G**/DFBS). Hydrogen atoms were removed except for the important ones to improve the clarity of pictures. Colour codes: light blue, white, red, blue, and ice-blue correspond to carbon, hydrogen, oxygen, nitrogen, and sodium, respectively. Colour codes of the isosurfaces: the weak-, the strong attractive-, and the strong repulsive interactions are in green, blue, and red, respectively. The black arrows indicate the isosurfaces discussed in the text. | |
Finally, we will address the question, whether the ion-pair (O−(3)–H+(4)) forms of the complexes 1S/RNa+ are really energetically preferred to their neutral counter-parts what might sound counterintuitive in the gas phase. The relaxed potential scans of a proton migration from N(5) to O−(3) were done for both isomers to answer this question. The scans show that the proton migration from the nitrogen atom to the oxygen atom is continuously endothermic (see Fig. S14–S19, ESI†). The relative energies of the neutral complexes are more than 18.0 kJ mol−1 above the energies of the ion-pair complexes. Hence, the spontaneous proton migration from O−(3) to N(5) occurred during the full optimization of the complexes and the ionic form of the O(3)–H(4) pair is preferred in the gas phase.
In summary, 1SNa+ and 1RNa+ are identified to be the most stable diastereoisomers and will be considered in the following discussion. Analysis of the stabilities of all computed isomers allows us to order all binding modes according to their contribution to the stabilization of the [tBuCQN·DNB-Leu·Na]+ complexes in the following order: the Coulomb interaction > the hydrogen bond ∼ the π−π interaction > van der Waals interaction. While the van der Waals interactions provide the smallest contribution to the stabilization, they make a decisive difference between the diastereoisomeric complexes and are responsible for the large potential of tBuCQN for the enantioselective recognition.
The effect of a counter ion
It was shown experimentally that changing a counter ion has a significant influence on the energy stabilities of the studied diastereoisomers.22 In the case of the lithium counter ions, enhancement in the separation ability of tBuCQN was observed compared to the sodium cations while using the potassium counter ions had an opposite effect. The 1SNa+ and 1RNa+ complexes are reoptimized with the Li and K counter ions in an attempt to see the relevant differences (see optimized structures in Fig. S6–S9, ESI†). The r1, r2, and r3 bond lengths are shortened in the lithium complexes compared to the sodium ones, especially in 1RLi+. The stronger interaction of Li+ with O(1), O(10), and O(11) is also confirmed by the respective isosurfaces that have a deeper blue colour (see Fig. 2b, 6b, Fig. S6b and S7b, ESI†). The O−(3)–H+(4) pair is affected slightly by the Na → Li exchange as well as the O(6)⋯H(7)–N(8) hydrogen bond and the π−π interaction. The more electronegative lithium absorbs more of the negative charge from the COO− group and it causes a lowering of the charge transfer from DNB-Leu to tBuCQN (Fig. 9). Consequently, the H(4) atom is bound tighter to N(5) and the O−(3)–H+(4) ionic interaction is weaker in 1S/RLi+ than in 1S/RNa+. The weakening is even more pronounced for 1RLi+ that is an opposite situation with respect to 1S/RNa+ in which the O−(3)–H+(4) is stronger in 1RNa+ than in 1SNa+. This can be also seen from a shift of the related spikes in the NCI plots ((sign(λ2)ρ is −0.072 and −0.071 for 1SLi+ and 1RLi+, respectively; see Fig. S10 and S11, ESI†). The impact of the Na → Li exchange on the interaction of the two alkyl groups is small. The r8 distance is elongated in the Li complexes with respect to the sodium ones.
![Dependence of charge transfers on electronegativity of the alkali metals (X = Li, Na, and K). The blue colour corresponds to [tBuCQN·DNB-S-Leu·X]+ and the red colour corresponds to [tBuCQN·DNB-R-Leu·X]+. Diamond is a partial charge of the alkali ion, cube is the amount of the charge transfer from DNB-S/R-Leu, and circle is the amount of the charge transfer to tBuCQN.](/image/article/2013/CP/c3cp44444a/c3cp44444a-f9.gif) |
| | Fig. 9 Dependence of charge transfers on electronegativity of the alkali metals (X = Li, Na, and K). The blue colour corresponds to [tBuCQN·DNB-S-Leu·X]+ and the red colour corresponds to [tBuCQN·DNB-R-Leu·X]+. Diamond is a partial charge of the alkali ion, cube is the amount of the charge transfer from DNB-S/R-Leu, and circle is the amount of the charge transfer to tBuCQN. | |
Similarly to 1RNa+, isosurfaces relating to the interaction of the two alkyl groups are not present in 1RLi+. Thus, the enantioselectivity is enhanced by the weakening of the O−(3)–H+(4) pair in 1SLi+ and 1RLi+ compared to those in 1SNa+ and 1RNa+ in that the weakening is more pronounced for 1RLi+. In the case of the potassium complexes (Fig. S8 and S9, ESI†), all three distances, r1, r2, and r3, are larger compared to the sodium and lithium complexes. The O−(3)–H+(4) pair interaction is weakened in 1SK+ with respect to 1SNa+, which is also indicated by the value of sign(λ2)ρ = −0.064 (Fig. S12, ESI†) of the related spike in the NCI plot. The stronger ionic pair interaction is found in 1RK+ than in 1SK+ and 1SNa+ (sign(λ2)ρ = −0.076; Fig. S13, ESI†). The lower electronegativity of K than Na and Li causes increasing charge transfer between the molecules with nearly single positive charge on K (Fig. 9). The Na → K exchange leads to the moderate weakening of the hydrogen bond in 1SK+ compared to 1SNa+ while strength of the hydrogen bond increases in its diastereoisomer 1RK+ with respect to 1SK+. On the other hand, the r8 distance is shortened in 1SK+ comparing with 1SNa+ and also 1RK+. The value of r9 then indicates that the π−π interaction is influenced only slightly by the Na → K exchange. The potassium counter ion makes the most important binding modes in the complex weaker in 1SK+ but stronger in 1RK+ similarly as in the sodium complexes. Consequently, the relative difference in the stabilities of 1SK+ and 1RK+ is smaller compared to the 1SNa+/1RNa+ pair and hence the enantioselectivity is poor using the potassium counter ion.
In summary, exchanging of the counter ion has an impact on three binding modes in each particular diastereoisomer. Only the π−π interaction does not show any significant differences with the exchanging of the counter ions and remains invariant to this.
4. Conclusions
The presented study shows a lower stability of the [tBuCQN·DNB-R-Leu·Na]+ complex in comparison with its diastereoisomer [tBuCQN·DNB-S-Leu·Na]+ in the gas phase mainly due to a weaker van der Waals interaction of the alkyl chains in [tBuCQN·DNB-R-Leu·Na]+. Furthermore, the O−(3)–H+(4) pair interaction is identified as the most stabilizing binding mode in [tBuCQN·DNB-S/R-Leu·Na]+ complexes followed by the other binding modes: a hydrogen bond and the π−π interaction. Replacing of the sodium cation with the more electronegative lithium cation causes a weakening of the O−(3)–H+(4) pair interaction, the hydrogen bond and also van der Waals interaction. The weakening of the binding modes is more pronounced in [tBuCQN·DNB-R-Leu·Li]+ than in [tBuCQN·DNB-S-Leu·Li]+. Hence, the energy difference between these two diastereoisomers is larger than for the sodium variants and thus the lithium ions are more suitable for the chiral recognition experiments. In contrast, the strength of the ionic pair interaction and the hydrogen bond is smaller in [tBuCQN·DNB-S-Leu·K]+ than in [tBuCQN·DNB-R-Leu·K]+. Therefore, the enantioselectivity is poor for the potassium complexes, despite the stronger vdW interaction in the potassium complexes. According to the work presented here, lithium appears to be the best counter ion in the chiral recognition within the [tBuCQN⋯DNB-S/R-Leu⋯X]+ (X = Li, Na, K) complexes, and this finding is consistent with the experimental mass spectrometry determinations.
Acknowledgements
The authors thank the late Dr habil. Detlef Schröder for providing computational time on the Horizoms computer cluster. This work was supported by the Ministry of Education of the Czech Republic (MSM0021620857). KAS acknowledges support from the National Science Foundation (CHE-0846310). KL acknowledges support from Operational Program Research and Development for Innovations – European Regional Development Fund (project CZ.1.05/2.1.00/03.0058) and from the Czech Science Foundation (project P206/12/1150).
Notes and references
- A. M. Rouhi, Chem. Eng. News, 2004, 82, 47 CrossRef.
- K. A. Schug, P. Fryčák, N. M. Maier and W. Lindner, Anal. Chem., 2005, 77, 3660 CrossRef CAS.
- Y. Okamoto and T. Ikai, Chem. Soc. Rev., 2008, 37, 2593 RSC.
- W. A. Tao and R. G. Cooks, Anal. Chem., 2003, 75, 25A CAS.
- S. Ahn, J. Ramirez, G. Grigorean and C. B. Lebrilla, J. Am. Soc. Mass Spectrom., 2001, 12, 278 CrossRef CAS.
- J. L. Seymour, F. Tureček, A. V. Malkov and P. Kočovský, J. Mass Spectrom., 2004, 39, 1044 CrossRef CAS.
- F. Gasparrini, M. Pierini, C. Villani, A. Filippi and M. Speranza, J. Am. Chem. Soc., 2008, 130, 522 CrossRef CAS.
- V. Ranc, V. Havlicek, P. Bednar and K. Lemr, Int. J. Mass Spectrom., 2009, 280, 213 Search PubMed.
- D. Scuderi, P. Maitre, F. Rondino, K. Le Barbu-Debus, V. Lepére and A. Zehnacker-Rentien, J. Phys. Chem. A, 2010, 114, 3306 CrossRef CAS.
- D. Scuderi, K. Le Barbu-Debus and A. Zehnacker, Phys. Chem. Chem. Phys., 2011, 13, 17916 RSC.
- K. A. Schug and W. Lindner, J. Sep. Sci., 2005, 28, 1932 CrossRef CAS.
- K. A. Schug, Comb. Chem. High Throughput Screening, 2007, 10, 301 Search PubMed.
-
A. Zehnacker, Chiral Recognition in the Gas Phase, Taylor & Francis/CRC Press, 2010, p. 181 Search PubMed.
- M. Bollini, R. A. Domaoal, V. V. Thakur, R. Gallardo-Macias, K. A. Spasov, K. S. Anderson and W. L. Jorgensen, J. Med. Chem., 2011, 54, 8582 Search PubMed.
- A. Czarna, B. Beck, S. Srivastava, G. M. Popowicz, S. Wolf, Y. Huang, M. Bista, T. A. Holak and A. Dömling, Angew. Chem., Int. Ed., 2010, 49, 5352 CrossRef CAS.
- E. Laine, C. Goncalves, J. C. Karst, A. Lesnard, S. Rault, W. J. Tang, T. E. Malliavin, D. Ladant and A. Blondel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11277 CrossRef CAS.
- T. C. Bruice, Chem. Rev., 2006, 106, 3119 CrossRef CAS.
- N. M. Maier, P. Franco and W. Lindner, J. Chromatogr., A, 2001, 906, 3 CrossRef CAS.
- M. Lämmerhofer and W. Lindner, J. Chromatogr., A, 1996, 741, 33 CrossRef.
- N. M. Maier, L. Nicoletti, M. Lämmerhofer and W. Lindner, Chirality, 1999, 11, 522 CrossRef CAS.
- N. M. Maier, S. Schefzick, G. M. Lombardo, M. Feliz, K. Rissanen, W. Lindner and K. B. Lipkowitz, J. Am. Chem. Soc., 2002, 124, 8611 CrossRef CAS.
- K. A. Schug, N. M. Maier and W. Linder, Chem. Commun., 2006, 414 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- J-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016 CrossRef CAS.
- B. I. Dunlap, J. Chem. Phys., 1983, 78, 3140 CrossRef CAS.
- B. I. Dunlap, THEOCHEM, 2000, 529, 37 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- Á. Vázquez-Mayagoitia, C. D. Sherrill, E. Aprà and B. G. Sumpter, J. Comput. Chem., 2010, 6, 727 Search PubMed.
- D. Josa, J. R. Otero and E. M. Cabaleiro Lago, Phys. Chem. Chem. Phys., 2011, 13, 21139 RSC.
- P. I. Nagy and P. W. Erhardt, J. Phys. Chem. B, 2012, 116, 5425 CrossRef CAS.
- A. B. González-Pérez, R. Álvarez, O. N. Faza, Á. R. de Lera and J. M. Aurrecoechea, Organometallics, 2012, 31, 2053 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theor. Comput., 2012, 8, 527 Search PubMed.
-
A. V. Marenich, C. J. Cramer and D. G. Truhlar, CM5PAC, University of Minnesota, Minneapolis, 2011 Search PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- J. P. Ritchie, J. Am. Chem. Soc., 1985, 107, 1829 CrossRef CAS.
- J. P. Ritchie and S. M. Bachrach, J. Comput. Chem., 1987, 8, 499–509 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS.
- M. O. Sinnokrot and C. D. Sherrill, J. Am. Chem. Soc., 2004, 126, 7690 CrossRef CAS.
- E. G. Hohenstein and C. D. Sherrill, J. Phys. Chem. A, 2009, 113, 878 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NCI plots, NBO analysis and xyz coordinates of the all optimized structures are posted. See DOI: 10.1039/c3cp44444a |
| ‡ Present address: Department of Chemical Research Support, Weizmann Institute of Science, Rehovot 76100, Israel. Tel: +972-8-934-6218. |
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a