 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sieving di-branched from mono-branched and linear alkanes using ZIF-8: experimental proof and theoretical explanation†
Alexandre F. P.
Ferreira
ab,
Marjo C.
Mittelmeijer-Hazeleger
b,
Miguel Angelo
Granato
a,
Vanessa F. Duarte
Martins
a,
Alírio E.
Rodrigues
a and
Gadi
Rothenberg
*b
aLaboratory of Separation and Reaction Engineering, Associate Laboratory LSRE/LCM, Department of Chemical Engineering, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, Porto, Portugal
bVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, Amsterdam, The Netherlands. E-mail: g.rothenberg@uva.nl; Web: http://hims.uva.nl/hcsc
First published on 2nd May 2013
Abstract
We study the adsorption equilibrium isotherms and differential heats of adsorption of hexane isomers on the zeolitic imidazolate framework ZIF-8. The studies are carried out at 373 K using a manometric set-up combined with a micro-calorimeter. We see that the Langmuir model describes well the isotherms for all four isomers (n-hexane, 2-methylpentane, 2,2-dimethylbutane and 2,3-dimethylbutane). The linear and mono-branched isomers adsorb well, but 2,2-dimethylbutane is totally excluded. Plotting the differential heat of adsorption against the loading shows an initial plateau for n-hexane and 2-methylpentane. This is followed by a slow rise, indicating adsorbate–adsorbate interactions. For the di-branched isomers the differential heat of adsorption decreases with loading. To gain further insight, we ran molecular simulations using the grand-canonical Monte Carlo approach. Comparing the simulation and the experimental results shows that the ZIF framework model requires blocking of the cages, since 2,2-dimethylbutane cannot fit through the sodalite-type windows. Practically speaking, this means that ZIF-8 is a highly promising candidate for enhancing gasoline octane numbers at 373 K, as it can separate 2,2-dimethylbutane and 2,3-dimethylbutane from 2-methylpentane. Our results prove the potential of ZIF-8 as a new adsorbent that can be employed in the upgrade of the Total Isomerization Process for the production of high octane number gasoline, by blending di-branched alkanes in the gasoline.
Introduction
Chemists and chemical engineers have long sought to use alkanes as feedstocks for fuels, plastics, solvents and pharmaceuticals. The major drawback has been that the bonds within alkane molecules are so strong that alkanes are generally unreactive. Yet, in recent years the development of new catalysts has opened new doors to the activation of resilient alkanes. A fine example is the hydrogenolysis of saturated hydrocarbons to generate high value chemicals and diesel-based hydrocarbons using more efficient catalysts, such as fibrous nano-silica supported ruthenium and tantalum hydride supported on MCM-41.1,2 Another important reaction is the alkylation of alkanes, which was discovered by Ipatieff and Pines in 1932.3 Today, this reaction is highly relevant in the production of Fischer–Tropsch diesel for the transport industry, and is still an active research field.4 Alkylation and isomerization are the main reactions for converting C4–C6 paraffins to high-octane, branched alkane gasoline additives. The alkanes in the gasoline fraction of crude oil are mainly linear or monobranched, and must be converted into highly branched isomers.Thus, the molecular separation of hexane isomers is a key step in gasoline enrichment and octane number enhancement. Traditionally, this is an area where both zeolites and metal–organic frameworks (MOFs) are well known.5–7 Especially zeolites are extensively studied in this context, due to their well-defined pores, the diameters of which are close to those of alkane molecules.7 The most studied structures for separating hexane isomers are ZSM-5,8–11 silicalite-1,11–22 zeolite beta,23–29 and mordenite.30–32 Compared to this, there are relatively few studies on applying MOFs for separating hexane isomers, mainly using UiO-66(zr),33 Zn(BDC)(DABCO)0.5,34,35 MIL-47,36 Cu(hfipbb)(H2hfipbb)0.5,37 and Zn(BDC)(4,4′-Bipy)0.5 (MOF-508).38
The problem is that cleanly separating fuel-grade hexanes remains a challenge, particularly where the cut between monobranched and dibranched isomers is concerned. Here we report the discovery that zeolitic imidazolate framework 8 (ZIF-8) can separate even 2,2-dimethylbutane from 2,3-dimethylbutane. Using a manometric set-up combined with a micro-calorimeter, we show that the sieving radius of ZIF-8 is between 5.8 and 6.3 Å. This value is much higher than the usually reported 5.3 Å.39 To the best of our knowledge, this is the first report that compares the adsorption isotherms of all four isomers. Moreover, we present the differential heats of adsorption of the four components, emphasizing the higher affinity of ZIF-8 for the linear and monobranched isomers, which was also translated by higher adsorption enthalpies. The gate opening effect, due to the flipping of the imidazole linkers, has been mimicked in molecular simulations using the well-established blocking strategy, which has the major advantage of being rather simple to implement. As our work deals with the fundamentals of adsorption on ZIFs, we give a short overview to enable all readers to place the results in the right context.
Zeolitic imidazolate frameworks (ZIFs) combine the properties of both zeolites and MOFs, such as high porosity, high surface area, thermal and chemical stability, and tuneable metal clusters and organic linkers.7,40,41 This gives a wide range of highly interesting structures. ZIF-8 has the formula Zn(mim)2 (where mim = 2-methylimidazole) with a sodalite-related type structure41 (see Fig. 1). At 3.4 Å diameter, the six-membered-ring pore windows of ZIF-8 are narrow, but the cages inside (11.4 Å diameter) are much larger.42 Thermogravimetric analyses of ZIF-8 showed a gradual weight-loss of 28.3% when the temperature was increased from 298 to 723 K, corresponding to the loss of guest species, then the structure was stable up to a temperature of 823 K,41 identical to the temperatures at which the structural collapse of zeolites takes place.43
![Optimised three-dimensional views of the ZIF-8 structure. Nitrogen, carbon and hydrogen atoms are shown in blue, light grey and white, respectively: (a) viewed along axis [001], (b) viewed along axis [111], (c) ZIF-8 sodalite-type cage, (d) SOD topology, and (e) 6-membered-ring window.](/image/article/2013/CP/c3cp44381g/c3cp44381g-f1.gif) | ||
| Fig. 1 Optimised three-dimensional views of the ZIF-8 structure. Nitrogen, carbon and hydrogen atoms are shown in blue, light grey and white, respectively: (a) viewed along axis [001], (b) viewed along axis [111], (c) ZIF-8 sodalite-type cage, (d) SOD topology, and (e) 6-membered-ring window. | ||
Several studies addressed the chromatographic separation of hexane isomers using ZIF-8 as a stationary phase. Chang et al.7 reported that branched alkanes eluted much earlier than their linear analogues on a ZIF-8 coated capillary. Moreover, 2,2-dimethylhexane eluted even earlier than hexane and heptane, despite its higher boiling point. In traditional stationary phases such as 5%-phenylmethylpolysiloxane, however, the elution followed the order of boiling points.7 Large-pore MOFs such as MOF-57 and MOF-50838 also showed the traditional elution sequence. Even though the diameter of the ZIF-8 pore window is only 3.4 Å, the framework is flexible, with no sharp sieving at 3.4 Å (methane can enter the pores).7,42,44 But bulkier branched alkanes (see Fig. 2) cannot pass through the narrow pore windows, giving a shorter retention time.7 Elsewhere, Lu and Hupp45 observed that a ZIF-8 sensor displayed some chemical selectivity for linear n-hexane over the bulkier cyclohexane,45 and Luebbers et al.46 reported that branched alkanes (except i-butane) were excluded from the pores. Thus, 2-methylbutane eluted very quickly, with practically no retention compared to n-pentane. Similar results were seen for 2-methylheptane.46 More recently, Peralta et al.39 reported the separation of linear and branched hexane isomers by breakthrough experiments of binary mixtures in three different ZIF materials. They concluded that ZIF-8 acts as a molecular sieve: linear alkanes diffuse freely into the pores, while mono-branched alkanes are adsorbed under strong diffusional limitation, and di-branched alkanes are excluded from the pores.39 They also concluded that the effective pore size of ZIF-8 is comparable to the kinetic diameter of mono-branched alkanes, 5.3 Å. Since this is higher than the formal pore size of 3.4 Å, it means that the flexibility of the pore aperture in ZIF-8 is much higher than anticipated.39
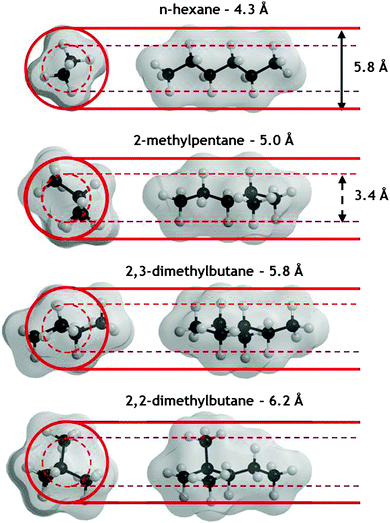 | ||
| Fig. 2 Three-dimensional views and kinetic diameters of hexane isomers: (a) n-hexane, (b) 2-methylpentane, (c) 2,3-dimethylbutane and (d) 2,2-dimethylbutane. Carbon and hydrogen atoms are in light grey and dark grey, respectively. The optimized structures were generated using ChemBio3D Ultra (CambridgeSoft). The kinetic diameters were reported by Gobin et al.53 | ||
Several molecular simulation studies address the adsorption properties of ZIFs for separating CH4, CO2, H2, and N2 binary mixtures,47–49 as well as ethane–ethene mixtures.50,51 The method used for methane adsorption on ZIF-8 reproduces well the experimental results, with slight overestimations.48,52
The above studies highlight the theoretical potential of ZIF-8 for separating hexane isomers. However, to prove this potential, we must obtain complete information on the adsorption equilibrium properties. To do this, we measured here the adsorption isotherms and differential heats of adsorption of n-hexane (n-C6), 2-methylpentane (2MP), 2,3-dimethylbutane (23DMB) and 2,2-dimethylbutane (22DMB) at 373 K. The experimental isotherms were modelled using the Langmuir model. Additionally, Henry's constants were calculated for the four components. The results prove the potential of ZIF-8 as a new adsorbent in the Total Isomerization Process for the production of high octane number gasoline, by blending di-branched alkanes in the gasoline.
Experimental
1. Materials and instrumentation
The adsorptives used were n-hexane (99+%, Merck), 2-methylpentane (99+%, Acros), 2,3-dimethylbutane (98+%, Acros) and 2,2-dimethylbutane (99+%, TCI). These liquids were vaporized, and then fed into the system without any further treatment. ZIF-8 was the commercially available Basolite® Z120054 (BASF). The sample (0.55 g) was evacuated for 6 h at 573 K, under a vacuum more than 2 × 10−6 mbar. N2 adsorption isotherms were determined at 77 K on a Surfer instrument (Thermo Scientific) and evaluated using the BET equation. The equilibrium isotherms and differential heats of adsorption were determined using a dedicated manometric system (this system is described in detail elsewhere9).Structure optimisations for Fig. 1 and 2 were generated using the ChemBio3D Ultra software (CambridgeSoft). Monte-Carlo simulations were carried out using the open source package RASPA 1.0 developed by Dubbeldam et al.55 This code was successfully applied in a large number of simulation studies.56–59 The simulations were performed using a 2 × 2 × 2 supercell of dimensions 33.986 × 33.986 × 33.986 Å3, typically using two million Monte Carlo steps.
2. Procedure for adsorption equilibrium isotherms and differential heats of adsorption measurement
The manometric introduction system was kept at 368 K, while the transfer line and the sample holder inside of the micro-calorimeter were kept at 373 K (Calvet C80, Setaram, see diagram and photo in Fig. 3). The experimental procedure comprises of measuring blanks, at 373 K, for the different vapors (correcting for the non-ideality of the vapors). Blank measurements were run by introducing a known amount of gas into the empty sample holder, and then measuring the pressure. During the adsorption measurement it was considered that the adsorption equilibrium was achieved if the pressure changed less than 0.03 kPa at a 100 min interval. | ||
| Fig. 3 Schematic and photograph of the experimental set-up, showing the micro-calorimeter in a thermostatic oven, the manometric introduction system, and the vacuum turbo-molecular pump. Photographs of detailed parts of equipment are presented in the ESI† – Fig. S4. | ||
Computational methods and models
1. Adsorption equilibrium and isotherm models
The structure of ZIF-8 consists of large cages connected by six-membered-ring windows in a sodalite topology.42 The windows have a diameter of approximately 0.34 nm.42 This suggests that there is only one adsorption site. Therefore, a Langmuir model (eqn (1)) can suffice for describing the adsorption: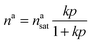 | (1) |
In the Henry's law region (low pressures) adsorption loading is directly proportional to the pressure (eqn (2), where KH denotes the Henry constant).
| q = KHp | (2) |
Since the Langmuir model is thermodynamically consistent, we obtain eqn (3):
| KH = nasatk | (3) |
2. Monte Carlo calculations
The hexane isomers are described using a united-atom model, in which the CHn groups are considered as a single interaction center without charge. In the Configurational Bias-Monte Carlo (CBMC) algorithm, chains are built from a first united atom (CHn is considered to be the united atom for the alkane molecules in this study) placed at a random position. The second atom is added and a harmonic bonding potential is used for the bond length. Chains are then grown segment by segment. The bond bending between three neighbouring beads is modelled by a harmonic cosine bending potential and the torsional angle is controlled by a Ryckaert-Bellemans potential. This bond length is fixed (1.53 Å).All non-bonded interactions are then described with the Lennard-Jones (LJ) 12–6 potential (eqn (4)),
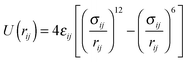 | (4) |
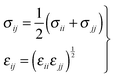 | (5) |
Tables 1 and 2 give the forcefield parameters. The LJ parameters of the ZIF-8 framework were taken from the UFF forcefield. Potential parameters for the adsorbates were taken from the TraPPE UA forcefield. Details of the force fields are found in Rappe et al.,60 Martin and Siepmann,61 and Calero et al.62 We use a truncated and shifted potential (cut-off radius = 12 Å) without tail corrections. Since the adsorbate molecules are non-polar, electrostatic interactions were ignored. Guo et al. simulated the adsorption of methane/hydrogen on ZIFs using the same set of parameters.47
| σ(Å) | ε/kB (K) | |
|---|---|---|
| United atom | ||
| CH3-sp3 | 3.76 | 108.00 |
| CH2-sp3 | 3.96 | 56.00 |
| CH-sp3 | 4.68 | 17.00 |
| C-sp3 | 0.80 | 6.38 |
| Framework | ||
| C | 3.43 | 52.84 |
| H | 2.57 | 22.14 |
| N | 3.26 | 34.72 |
| O | 3.12 | 30.19 |
| Zn | 2.46 | 62.40 |
| Bond |
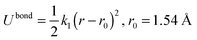
|
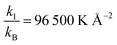
|
|||
| Bend |
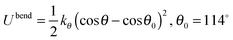
|
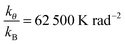
|
|||
| Torsion |
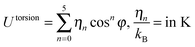
|
||||
| η 0 | η 1 | η 2 | η 3 | η 4 | η 5 |
| 1204.654 | 1947.740 | −357.854 | −1944.666 | 715.690 | −1565.572 |
3. Framework model and blocking of sodalite cages
Because of the presence of long linkers rather than bridging O atoms, the pore cages in ZIF-8 are about 11.6 Å. They are connected via small apertures with a diameter of 3.4 Å. Thus, even though a hexane isomer fits in the cage, it cannot enter it. As the CBMC method randomly grows a guest molecule atom by atom inside the cavities, hexane isomer molecules could actually be “loaded” into these cages. To prevent this, we blocked the cages artificially using the procedure outlined by Dubbeldam et al.63 Omitting this blocking could lead to false higher loadings, especially at low temperatures and/or high pressures (the favourable conditions for high adsorption loading).Results
1. Nitrogen equilibrium adsorption data
The nitrogen adsorption isotherm is presented in Fig. 4. We see that the calculated micropore volume (Vmic/cm3 g−1) is 0.73. This volume was obtained by applying the Dubinin–Radushkevich (DR) equation to the N2 equilibrium data.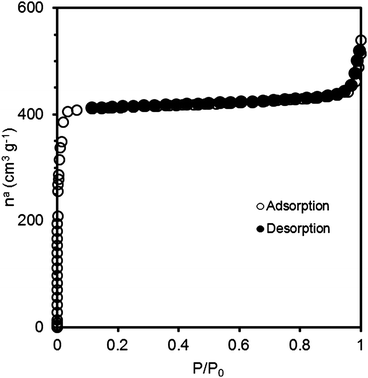 | ||
| Fig. 4 N2 adsorption isotherm on ZIF-8 at 77 K. | ||
Applying the BET equation to the data gives a surface area value of 1800 m2 g−1. This value affirms the manufacturer's specification of 1300–1800 m2 g−1.54 It also confirms that the activation procedure has regenerated the sample to its original state, removing any component (e.g. water) from the pores.
2. Hexane isomer equilibrium adsorption data
We then measured the adsorption equilibrium data of n-C6, 2-MP, 2,3-DMB and 2,2-DMB on ZIF-8 at 373 K, for a pressure range from 0.01 kPa to 100 kPa (see Fig. 5).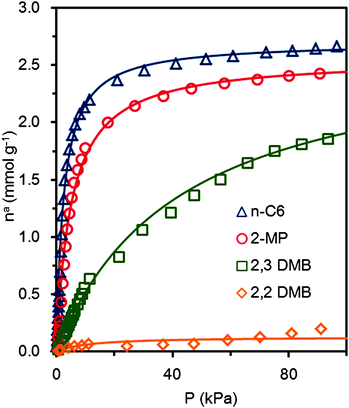 | ||
| Fig. 5 Hexane isomer adsorption isotherms, on ZIF-8, at 373 K: △: n-hexane, ○: 2-methylpentane, □: 2,3-dimethylbutane, and ⋄: 2,2-dimethylbutane. The curves show the Langmuir model fit for each isotherm, calculated using eqn (1). | ||
Fig. 5 shows that ZIF-8 adsorbs n-hexane selectively. However, 2-methylpentane and 2,3-dimethylbutane present similar adsorption saturation loading, but with a lower Langmuir equilibrium constant (about half for the mono-branched isomer and twenty times lower for the di-branched isomer). Thus, we consider that the maximum capacity of the ZIF-8 cages is around ∼2.7 mmol g−1 (7.37 molecules (u.c.)−1). The 2,2-dimethylbutane is almost excluded. Langmuir's model describes well the isotherms for all four isomers (see parameters in Table 3). Plotting the differential heat of adsorption against the loading (see Fig. 6) gives an initial plateau for n-hexane at around 42–44 kJ mol−1. This plateau is followed by a slow rise until about 60 kJ mol−1. We attribute this to increasing adsorbate–adsorbate interactions.
| Parameters | ||
|---|---|---|
| n asat* [mmol g−1] | K [kPa−1] | |
| n-Hexane | 2.70 | 0.40 |
| 2-Methylpentane | 2.57 | 0.20 |
| 2,3-Dimethylbutane | 2.72 | 0.02 |
| 2,2-Dimethylbutane | 0.13 | 0.09 |
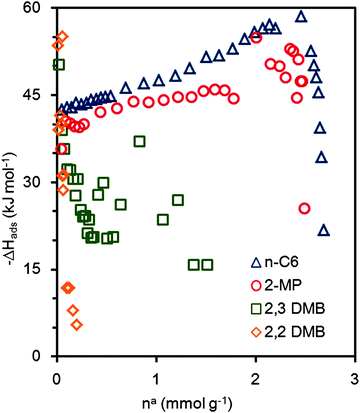 | ||
| Fig. 6 Differential heats of adsorption for hexane isomers on ZIF-8 at 373 K: △: n-hexane, ○: 2-methylpentane, □: 2,3-dimethylbutane, and ⋄: 2,2-dimethylbutane. | ||
The differential heats of adsorption of 2-methylpentane present similar trends to n-hexane. For the di-branched isomers the differential heat of adsorption decreases with loading. This decrease is sharper for the di-branched isomer. Overall, ZIF-8 shows a promising behaviour at 373 K for gasoline octane number enhancement, since it presents selectivity between 2-methylpentane and 2,3-dimethylbutane.
3. Henry constants
Table 4 presents the Henry constant values for the four isomers. These values were calculated using the slope of the line passing by the first data points of each isotherm (at least 8 points). This approach generally gives a good estimation for the Henry constant values. The values were compared with the value of the product nasat × k, to check the thermodynamic consistency of the model parameters. These results show that n-hexane has the highest affinity towards the adsorbent.| K H [mmol g−1 kPa−1] | n asat k [mmol g−1 kPa−1] | |
|---|---|---|
| n-Hexane | 0.56 | 1.07 |
| 2-Methylpentane | 0.26 | 0.51 |
| 2,3-Dimethylbutane | 0.06 | 0.06 |
| 2,2-Dimethylbutane | 0.005 | 0.011 |
4. Molecular simulations
The molecular simulations were ran using the CBMC technique in the μVT ensemble, where the chemical potential, temperature and volume are kept constant.As Fig. 7a shows, if no blocking is used, all isomers quickly saturate the adsorbent. Almost no selectivity is observed. A zoom-in of the region with pressures up to 20 kPa is included in the ESI† (Fig. S2). The maximum saturation is about 2.9 mmol g−1, corresponding to eight molecules per unit cell. When appropriate blocking is applied, the simulations correctly reproduce the experimental isotherms (see Fig. S3 ESI†). The blocking strategy comprises rejection of all Monte Carlo moves that attempt to insert a molecule inside a certain radius. Fig. 8 shows a snapshot of the maximum adsorption loading of n-hexane in ZIF-8. The most favourable adsorption site for the n-hexane molecules is inside the cages. No adsorption occurs at the windows.
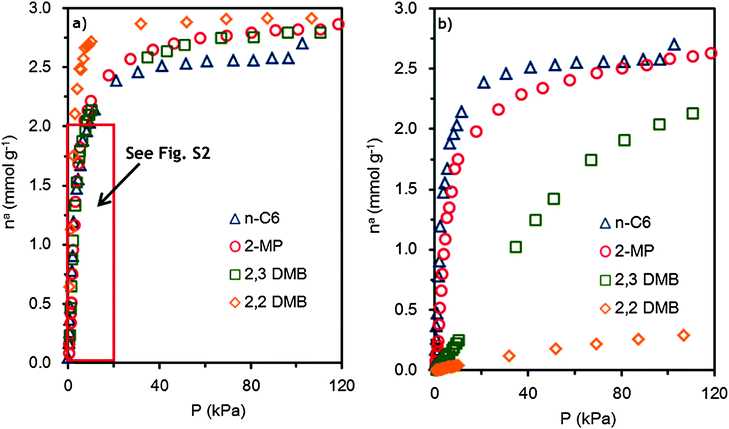 | ||
| Fig. 7 Molecular simulation of the hexane isomer adsorption isotherms on ZIF-8 at 373 K: (a) without blocking and (b) with blocking. (△: n-hexane, ○: 2-methylpentane, □: 2,3-dimethylbutane, and ⋄: 2,2-dimethylbutane). | ||
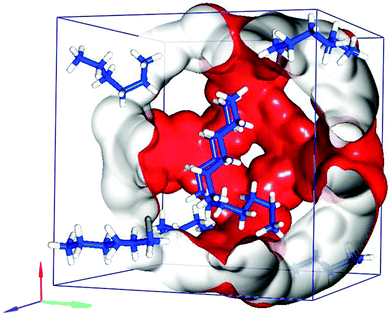 | ||
| Fig. 8 Snapshot of adsorbed n-hexane in one ZIF-8 unit cell at 102 kPa. | ||
Discussion
The capacity of n-hexane is 7.3 molecules per unit cell, corresponding to 22.11 carbon atoms per cage. This is in good agreement with the results of Bux et al.,50 who reported a loading of 22 carbon atoms per cage for ethane in ZIF-8 at 298 K. Furthermore, applying the Van't Hoff equation to the values reported by Luebbers et al. (Table 1 in ref. 41) we get a Henry constant of 0.82 mmol g−1 kPa−1 for n-hexane at 373 K.46 This agrees with our own results (see Table 4). Using the Van't Hoff equation we can also derive the enthalpy of adsorption for n-hexane in ZIF-8, which equals 41.4 kJ mol−1. This differs slightly from the value reported by Luebbers et al. (37.5 kJ mol−1). Nevertheless, both values are close to our experimental result, measured by microcalorimetry, of 42.4 kJ mol−1.Peralta et al.39 concluded, by means of binary vapour phase breakthrough experiments of n-hexane and 3-methylpentane, that the mono-branched isomer has a slow diffusion (three orders of magnitude lower than the n-hexane) and is also thermodynamically less favoured, with adsorption constant half of the linear isomer. The parameters obtained by fitting our data to a Langmuir isotherm model give an adsorption constant for n-hexane of 0.40 kPa−1 and for 2-methylpentane of 0.20 kPa−1. These values are in good agreement with the conclusions of Peralta et al.39 Although we haven't performed up-take experiments to assess the diffusion constants, when we compare the equilibration times required for both isomers (see Tables in the ESI†) we see that on average the 2-methylpentane takes about four times longer per point than the n-hexane. We attribute this to the slower diffusion of the mono-branched isomer.
The effective adsorption of 2,3-dimethylbutane and exclusion of 2,2-dimethylbutane prove that the effective pore size of ZIF-8 is about 5.8 Å, up to a maximum of 6.3 Å. This is at least half an Ångström higher than the value presented by Peralta et al.,39 and almost double of the reported window size (3.4 Å).
Table 4 shows that the Langmuir model over-predicts the adsorption capacity at very low pressures for the n-hexane and 2-methylpentane. This is due to an inflection on the isotherm of those two isomers that happens at about 1 kPa (see Fig. S1 in ESI†). This might be due to a first step of adsorption in the cage windows at the surface of the crystals, since the inflection occurs at about 0.25–0.5 mmol g−1, which is similar to the total amount of 2,2-dimethylbutane adsorbed that can be attributed to surface adsorption. Practically speaking, the Langmuir model represents well the adsorption equilibrium of the four isomers, since for pressures above 2 kPa the calculated adsorbed amount is close to the experimental value (note that in the regeneration step of the Total Isomerization Process, such low partial pressures are probably irrelevant).
The very low (yet still measurable) adsorption capacity of 2,2-dimethylbutane is attributed to “outer surface” adsorption. These molecules are too bulky to enter the cages, but it is possible that the ethyl group “anchors” in the window, leading to a certain amount of “external surface” adsorption that will depend on the crystallite size.
As we explained above, when no blocking is applied, the simulations show a slightly lower capacity for the linear isomer. The more compact 2,2-dimethylbutane presents the highest adsorption capacity. This is easily understood as the ZIF-8 cages are big when compared with the adsorbate molecules, and therefore, the packing effects (entropic) favour the di-branched isomers. This kind of effect was already observed in large-pore zeolites, such as mordenite.64 Nevertheless, we reasoned from experimental vs. simulation results that the adsorption behaviour of hexane isomers in ZIF-8 is not controlled by the cage accessible volume, but by the windows size and its “gate-opening” effect. This “gate-opening” effect is characteristic of some ZIF materials, as reported by van den Bergh et al.65,66 To reproduce the experimental results by CBMC simulations, we designed an appropriate blocking strategy. Blocking strategies are well known and simple to be employed. We obtained a good agreement between experimental and simulation results (see Fig. S3, ESI†). Note that for simulation of the n-hexane isotherm no blocking was necessary. This might be because of the smaller kinetic diameter of n-hexane, therefore the gate-opening effect requires very low pressures for the n-hexane or even its adsorption is not affected by the window size at all.
Conclusions
From the adsorption equilibrium isotherms we conclude that ZIF-8 is selective for n-hexane and totally excludes the bulky 2,2-dimethylbutane. Moreover, our CBMC simulations with appropriate blocking predict well the adsorption of n-hexane on ZIF-8.67 ZIF-8 is a promising material for separating both linear and mono-branched alkanes from the di-branched ones, and therefore has a great potential for improving the Total Isomerization Process to obtain high-octane gasoline.Nomenclature
| ΔHads | Heat of adsorption |
| k | Adsorption equilibrium constant |
| K 1, Kθ | Constants related to the bonded interactions: bond stretching and bond bending, respectively |
| K B | Boltzmann's constant |
| K H | Henry constant |
| n a | Loading (adsorbate concentration in the particles) |
| p | Pressure (adsorbate concentration in the gas phase) |
| r | Bond length |
| T | Temperature |
| U | Potential energy related to the bond, bend and torsion potentials, respectively |
| V | Volume |
Greek letters
| ε | Characteristic energy in LJ potential |
| ϕ | Torsion angle |
| η | Constants related to torsional configurations |
| μ | Chemical potential |
| θ | Bending angle |
| σ | Characteristic distance in LJ potential |
Subscripts
| ads | adsorption |
| sat | saturation |
Superscripts
| bend | Related to bending potential |
| bond | Related to bonding potential |
| torsion | Related to torsion potential |
Acknowledgements
M.A.G. the Fundação para a Ciência e a Tecnologia (FCT) for financial support (post-doctoral grant SFRH/BPD/47432/2008). V.D.M. acknowledges a scholarship from project PTDCEQU/ERQ/104413/2008. This work was partially supported by projects PTDC/EQU/ERQ/104413/2008 and Pest-C/EQB/LA0020/2011, financed by FEDER through COMPETE—Programa Operacional Factores de Competitividade and by FCT—Fundação para a Ciência e a Tecnologia.Notes and references
- A. Fihri, M. Bouhrara, U. Patil, D. Cha, Y. Saih and V. Polshettiwar, ACS Catal., 2012, 2, 1425–1431 Search PubMed.
- V. Polshettiwar, F. A. Pasha, A. De Mallmann, S. Norsic, J. Thivolle-Cazat and J.-M. Basset, ChemCatChem, 2012, 4, 363–369 Search PubMed.
- G. Egloff and G. Hulla, Chem. Rev., 1945, 37, 323–399 Search PubMed.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- Contents and Chemical Science, ed. J. Long and O. Yaghi, The Royal Society of Chemistry, 2009 Search PubMed.
- N. Chang, Z. Y. Gu and X. P. Yan, J. Am. Chem. Soc., 2010, 132, 13645–13647 CrossRef CAS.
- A. F. P. Ferreira, M. Mittelmeijer and A. Bliek, Stud. Surf. Sci. Catal., 2004, 154, 1971–1977 Search PubMed.
- A. F. P. Ferreira, M. C. Mittelmeijer-Hazeleger and A. Bliek, Microporous Mesoporous Mater., 2006, 91, 47–52 CrossRef CAS.
- A. F. P. Ferreira, M. C. Mittelmeijer-Hazeleger and A. Bliek, Adsorption, 2007, 13, 105–114 Search PubMed.
- A. F. P. Ferreira, M. C. Mittelmeijer-Hazeleger, A. Bliek and J. A. Moulijn, Microporous Mesoporous Mater., 2008, 111, 171–177 Search PubMed.
- A. C. Dubreuil, E. Jolimaitre, M. Tayakout-Fayolle and A. Methivier, Ind. Eng. Chem. Res., 2008, 47, 2386–2390 Search PubMed.
- E. Jolimaitre, K. Ragil, M. Tayakout-Fayolle and C. Jallut, AIChE J., 2002, 48, 1927–1937 CrossRef CAS.
- E. Jolimaitre, M. Tayakout-Fayolle, C. Jallut and K. Ragil, Ind. Eng. Chem. Res., 2001, 40, 914–926 Search PubMed.
- K. Lettat, E. Jolimaitre, M. Tayakout and D. Tondeur, AIChE J., 2011, 57, 319–332 Search PubMed.
- T. J. H. Vlugt, W. Zhu, F. Kapteijn, J. A. Moulijn, B. Smit and R. Krishna, J. Am. Chem. Soc., 1998, 120, 5599–5600 CrossRef CAS.
- W. Zhu, F. Kapteijn and J. A. Moulijn, Phys. Chem. Chem. Phys., 2000, 2, 1989–1995 RSC.
- W. Zhu, F. Kapteijn and J. A. Moulijn, Adsorption, 2000, 6, 159–167 CrossRef CAS.
- W. Zhu, F. Kapteijn and J. A. Moulijn, Microporous Mesoporous Mater., 2001, 47, 157–171 CrossRef CAS.
- W. Zhu, F. Kapteijn and J. A. Moulijn, Sep. Purif. Technol., 2003, 32, 223–230 CrossRef CAS.
- W. Zhu, F. Kapteijn, B. van der Linden and J. A. Moulijn, Phys. Chem. Chem. Phys., 2001, 3, 1755–1761 RSC.
- W. Zhu, J. M. van de Graaf, L. J. P. van den Broeke, F. Kapteijn and J. A. Moulijn, Ind. Eng. Chem. Res., 1998, 37, 1934–1942 CrossRef CAS.
- P. S. Barcia, A. Ferreira, J. Gascon, S. Aguado, J. A. C. Silva, A. E. Rodrigues and F. Kapteijn, Microporous Mesoporous Mater., 2010, 128, 194–202 CrossRef CAS.
- P. S. Barcia, J. A. C. Silva and A. E. Rodrigues, Microporous Mesoporous Mater., 2005, 79, 145–163 CrossRef CAS.
- P. S. Barcia, J. A. C. Silva and A. E. Rodrigues, Ind. Eng. Chem. Res., 2006, 45, 4316–4328 CrossRef CAS.
- P. S. Barcia, J. A. C. Silva and A. E. Rodrigues, AIChE J., 2007, 53, 1970–1981 CrossRef CAS.
- P. S. Barcia, J. A. C. Silva and A. E. Rodrigues, Adsorpt. Sci. Technol., 2007, 25, 169–183 Search PubMed.
- P. S. Barcia, J. A. C. Silva and A. E. Rodrigues, Energy Fuels, 2010, 24, 5116–5130 Search PubMed.
- E. Garcia-Perez, P. S. Barcia, J. A. C. Silva, A. E. Rodrigues and S. Calero, Theor. Chem. Acc., 2011, 128, 695–703 Search PubMed.
- K. Huddersman and M. Klimczyk, AIChE J., 1996, 42, 405–408 Search PubMed.
- K. Huddersman, AIChE J., 1996, 42, 2990–2992 Search PubMed.
- J. M. van Baten and R. Krishna, Microporous Mesoporous Mater., 2005, 84, 179–191 CrossRef CAS.
- P. S. Barcia, D. Guimaraes, P. A. P. Mendes, J. A. C. Silva, V. Guillerm, H. Chevreau, C. Serre and A. E. Rodrigues, Microporous Mesoporous Mater., 2011, 139, 67–73 CrossRef CAS.
- P. S. Barcia, F. Zapata, J. A. C. Silva, A. E. Rodrigues and B. L. Chen, J. Phys. Chem. B, 2007, 111, 6101–6103 CrossRef.
- D. Dubbeldam, C. J. Galvin, K. S. Walton, D. E. Ellis and R. Q. Snurr, J. Am. Chem. Soc., 2008, 130, 10884–+ CrossRef CAS.
- V. Finsy, S. Calero, E. Garcia-Perez, P. J. Merkling, G. Vedts, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Phys. Chem. Chem. Phys., 2009, 11, 3515–3521 RSC.
- L. Pan, D. H. Olson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616–619 CrossRef CAS.
- B. L. Chen, C. D. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS.
- D. Peralta, G. Chaplais, A. Simon-Masseron, K. Barthelet and G. D. Pirngruber, Ind. Eng. Chem. Res., 2012, 51, 4692–4702 Search PubMed.
- A. P. Cote and O. M. Yaghi, Abstr. Paper. Am. Chem. Soc., 2007, 233, 61 Search PubMed.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- H. Bux, F. Y. Liang, Y. S. Li, J. Cravillon, M. Wiebcke and J. Caro, J. Am. Chem. Soc., 2009, 131, 16000–16001 CrossRef CAS.
- G. Cruciani, J. Phys. Chem. Solids, 2006, 67, 1973–1994 CrossRef CAS.
- W. Zhou, H. Wu, T. J. Udovic, J. J. Rush and T. Yildirim, J. Phys. Chem. A, 2008, 112, 12602–12606 CrossRef CAS.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832–7833 CrossRef CAS.
- M. T. Luebbers, T. J. Wu, L. J. Shen and R. I. Masel, Langmuir, 2010, 26, 15625–15633 CrossRef CAS.
- H. C. Guo, F. Shi, Z. F. Ma and X. Q. Liu, J. Phys. Chem. C, 2010, 114, 12158–12165 CrossRef CAS.
- J. Perez-Pellitero, H. Amrouche, F. R. Siperstein, G. Pirngruber, C. Nieto-Draghi, G. Chaplais, A. Simon-Masseron, D. Bazer-Bachi, D. Peralta and N. Bats, Chem.–Eur. J., 2010, 16, 1560–1571 CrossRef CAS.
- A. Battisti, S. Taioli and G. Garberoglio, Microporous Mesoporous Mater., 2011, 143, 46–53 CrossRef CAS.
- H. Bux, C. Chmelik, R. Krishna and J. Caro, J. Membr. Sci., 2011, 369, 284–289 CrossRef CAS.
- H. Bux, C. Chmelik, J. M. van Baten, R. Krishna and J. Caro, Adv. Mater., 2010, 22, 4741–4743 CrossRef CAS.
- W. Zhou, H. Wu, M. R. Hartman and T. Yildirim, J. Phys. Chem. C, 2007, 111, 16131–16137 CrossRef CAS.
- O. C. Gobin, S. J. Reitmeier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 1171–1179 CrossRef CAS.
- 2-Methylimidazole zinc salt, MSDS sheet (online). Sigma-Aldrich Quimica SA, Madrid, Spain.
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, RASPA 1.0: Molecular Software Package for Adsorption and Diffusion in (Flexible) Nanoporous Materials, 2009 Search PubMed.
- S. Calero, A. Martin-Calvo, S. Hamad and E. Garcia-Perez, Chem. Commun., 2011, 47, 508–510 RSC.
- J. M. Castillo, T. J. H. Vlugt and S. Calero, J. Phys. Chem. C, 2008, 112, 15934–15939 CrossRef CAS.
- J. M. Castillo, T. J. H. Vlugt and S. Calero, J. Phys. Chem. C, 2009, 113, 20869–20874 CrossRef CAS.
- A. García-Sánchez, C. O. Ania, J. B. Parra, D. Dubbeldam, T. J. H. Vlugt, R. Krishna and S. Calero, J. Phys. Chem. C, 2009, 113, 8814–8820 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- S. Calero, D. Dubbeldam, R. Krishna, B. Smit, T. J. H. Vlugt, J. F. M. Denayer, J. A. Martens and T. L. M. Maesen, J. Am. Chem. Soc., 2004, 126, 11377–11386 CrossRef CAS.
- D. Dubbeldam, E. Beerdsen, T. J. H. Vlugt and B. Smit, J. Chem. Phys., 2005, 122, 224712 CrossRef CAS.
- M. Schenk, S. Calero, T. L. M. Maesen, T. J. H. Vlugt, L. L. van Benthem, M. G. Verbeek, B. Schnell and B. Smit, J. Catal., 2003, 214, 88–99 Search PubMed.
- C. Gucuyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef.
- J. van den Bergh, C. Gucuyener, E. A. Pidko, E. J. M. Hensen, J. Gascon and F. Kapteijn, Chem.–Eur. J., 2011, 17, 8832–8840 CrossRef CAS.
- E. J. Ras, M. J. Louwerse, M. C. Mittelmeijer-Hazeleger and G. Rothenberg, Phys. Chem. Chem. Phys., 2013, 15, 4436–4443 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp44381g |
| This journal is © the Owner Societies 2013 |