 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemically-synthesised, atomically-precise gold clusters deposited and activated on titania†
David P.
Anderson
a,
Jason F.
Alvino
b,
Alexander
Gentleman
b,
Hassan Al
Qahtani
c,
Lars
Thomsen
d,
Matthew I. J.
Polson
a,
Gregory F.
Metha
*b,
Vladimir B.
Golovko
*a and
Gunther G.
Andersson
*c
aThe MacDiarmid Institute for Advanced Materials and Nanotechnology, Department of Chemistry, University of Canterbury, Christchurch 8140, New Zealand. E-mail: vladimir.golovko@canterbury.ac.nz
bDepartment of Chemistry, University of Adelaide, 5005, Australia. E-mail: greg.metha@adelaide.edu.au
cFlinders Centre for NanoScale Science and Technology, Flinders University, Adelaide SA 5001, Australia. E-mail: gunther.andersson@flinders.edu.au
dAustralian Synchrotron, 800 Blackburn Road, Clayton Vic-3168, Australia
First published on 17th January 2013
Abstract
Synchrotron XPS was used to investigate a series of chemically-synthesised, atomically-precise gold clusters Aun(PPh3)y (n = 8, 9, 11 and 101, with y depending on cluster size) immobilized on titania nanoparticles. The gold clusters were washed with toluene at 100 °C or calcined at 200 °C to remove the organic ligand. From the position of the Au 4f7/2 peak it is concluded that cluster size is not altered through the deposition. From the analysis of the phosphorous spectra, it can be concluded that the applied heat treatment removes the organic ligands. Washing and calcination leads to partial oxidation and partial agglomeration of the clusters. Oxidation of the clusters is most likely due to the interaction of the cluster core with the oxygen of the titania surface after removal of ligands. The position of the Au 4f7/2 peak indicates that the size of the agglomerated clusters is still smaller than that of Au101.
Introduction
Fabrication of support-immobilized nanoparticles as model systems1,2 in heterogeneous catalysis follows two distinct routes. The first route relies on the classical surface science UHV techniques. In this case, metal nanoparticles are created on flat supports either via gas phase delivery and support-assisted self-assembly3 of metal atoms by evaporation of the metal4–6 or by the deposition of size-selected clusters formed in the gas phase after the evaporation of the metal.7–9 The second route involves chemical synthesis of nanoparticles or their precursors followed by their deposition onto the support. This route offers the huge benefit of easy scale-up and testing of the fabricated materials under industrially relevant conditions. Generally, there are three alternative methods for the chemical catalyst fabrication route, with varying degree of control over the precision with which the nanoparticles are created. The simple chemical methods aim to control the growth of nanoparticles on various supports via reduction/decomposition of simple precursors, such as simple metal salts or simple organometallic compounds.10 Improved control can be achieved via the immobilization of pre-made metal ion containing templates, such as metal ion containing dendrimers,11 onto supports followed by careful reduction/decomposition to form well defined nanoparticles. Alternatively, the metal core (containing metal–metal bonds) of the nanoparticle can be formed during the synthesis stage (i.e. in the liquid phase cf. on the support surface) with an excellent degree of control over the size and chemical composition. Liquid phase synthesis of gold colloids with narrow metal core size distributions, stabilized by capping groups dates back to the 19th century studies by Faraday.12Immobilization of colloids and clusters onto supports is an important step in the fabrication of heterogeneous catalysts. The post-fabrication activation of such catalysts by calcination13–17 or washing18 could afford nanoparticles, from which some or all ligands have been removed and where the size is defined by the specific precursor (colloid or cluster). However, in some cases catalytic activity is attributed to intact clusters on the support, i.e. clusters from which ligands have not been removed.19 Detailed studies of the effects of the activation processes on the morphology and electronic properties of the support-immobilized nanoparticles are required to understand the nature of such catalysts. Importantly, support-immobilization of atomically-precise chemically-synthesised clusters offers an opportunity to control the active site of the heterogeneous catalyst with an ultimate precision allowing correlation of any observed catalytic reactivity with the structural and electronic properties of the key ingredients – exactly defined metal moieties. Thus, detailed understanding of the deposition of atomically precise gold clusters and the effect of activation processes is important for the development of better strategies for fabrication of heterogeneous catalysts.
Titania is a particularly important support material in the field of gold nanoparticle-based heterogeneous catalysts.3 Surprisingly, fabrication and activation of catalysts using chemically-synthesised, atomically precise gold clusters supported on titania is practically unexplored.20 Thus, for the purpose of this study we have focused our efforts on gold clusters supported on TiO2 P-25 Aeroxide – a commercially available material which is often used as a standard in the field of TiO2-focused research.
Gold particles with a size reduced below 5 nanometres are known to be catalytically active.21,22 Moreover, it was demonstrated that the onset of catalytic activity of gold coincides with the loss of metallic properties of Au nanoparticles at around 1.5–2 nm. This size-threshold is established for both naked clusters prepared under UHV conditions23 and chemically synthesised,14 well-defined metal nanoparticles immobilized onto inert supports.
More recently, there have been reports that the reactivity properties can become even more dramatic when the particle size is less than 1 nm, where each particle can be considered as a nanocluster (or cluster). There is evidence of excellent catalytic performance of specific, size-selected clusters produced in the gas phase. For example, the CO oxidation by Au8 clusters deposited on MgO surfaces has been experimentally shown to occur at 150 K,9 whereas smaller clusters are inert. Hutchings and co-workers have demonstrated that a tiny amount of gold nanoclusters consisting of ca. 10 atoms is responsible for high CO-oxidation activity of a catalyst containing a majority of gold in the form of larger particles immobilized on iron-oxide.24 Similarly, Flytzani-Stephanopoulos and co-workers have shown that sub-1 nm gold particles on ceria are highly active in the methanol-steam-reforming and water-gas-shift reactions, compared to effectively inactive particles larger than 3 nm in size.25,26 Finally, chemically made, atomically-precise gold clusters stabilized by thiols (i.e. strongly bonding ligands) have very recently been shown to be active in reactions of oxidation and hydrogenation.27–29
Whilst there is general agreement that the size and the electronic structure of the active components of the nanoparticles plays a critical role in the catalytic process, systematic studies revealing the electronic structure of chemically-synthesized gold clusters used in catalysis are still very rare. For this reason, electronic spectroscopy, such as XPS, is an important tool in identifying the electronic effects at play.30 In particular, many studies have focussed on the 4f7/2 orbital of gold as it provides a convenient and sensitive measure of the electronic state of gold. In the following five paragraphs we will give an overview of how the binding energy of the core electrons determined by XPS informs about the size and chemical state of the deposited clusters. This overview is essential for the interpretation of the XPS spectra. However, the reader could also proceed to the end of this section where a summary is provided.
Firstly, the oxidation state plays a major role in the peak position. The literal gold standard is bulk gold (i.e. Au0), in which the 4f7/2 peak is well known to appear at 84.0 eV with a 3.67 eV separation to the 4f5/2 peak. Upon complete oxidation to Au2O3, the 4f7/2 peak shifts to 85.8–86.2 eV, depending upon the substrate;31,32 the thickness of gold oxide layer is estimated to be between 0.75 nm31 and 3.3 nm,32 providing the 4f7/2 peak position values for a macroscopic layer of gold oxide surface (cf. nanoparticles of Au2O3). Secondly, initial and final states effects also determine the peak position as well as the full-width-half-maximum of the peaks, both depending on the element forming the cluster, its size and its interaction with the substrate.33 Changes in the valence electron structure resulting from reduced size of the cluster (cf. the bulk metal) and changes due to the interaction with the substrate contribute to the initial state effects.33 After excitation, the metal nanocluster can stay in a charged state for a certain period of time, which often depends on the size of the cluster and the type of the substrate. This is often referred to as a final state effect.33 Therefore, the peak position and width can provide information that is of fundamental importance for understanding the electronic structure of metal nano-clusters, whether they are prepared via gas phase deposition or are chemically synthesised. However, interpreting changes of peak positions as an initial or final state effect based on experimental data is very difficult for gold as the Auger lines are rather weak.33,34 Thus, in the present work only changes in peak positions will be considered and not interpreted as initial or final state effects.
Over the last decade there have been a number of studies of sub-monolayer gold films evaporated onto various substrates and of chemically synthesised clusters in various matrices. A large variety of results was reported. The difficulty with finding a unifying thread between these studies is the uncertainty in particle size, particularly below 1 nm, where exact size determination is difficult. We provide a thorough, though not exhaustive, description of previous work using XPS to determine key properties of ultra-small gold particles/clusters. It is ordered according to the specific way in which the materials have been fabricated. We will start with gold nanoparticles within the ultra-small (sub-2 nm) particle size-regime made without precise control of the particle sizes; followed by atomically precise naked gold clusters made under UHV conditions, size-selected and soft-landed onto specific support material; and finally discuss the atomically-precise, chemically-synthesised ligand protected gold clusters deposited onto a specific support from solution.
Büttner et al. evaporated gold onto pristine HOPG and defect-rich HOPG following bombardment with Ar ions. No peak shifts were found on the pristine surface, whereas gold on the defect-rich HOPG shows peak shifts of up to +1.5 eV, multiple peaks and peak broadening.4 The authors attribute the peak shift and broadening to the preferential formation of gold particles at the defect sites which are found in a much higher number on the HOPG exposed to Ar bombardment. Kitsudo et al. evaporated gold onto graphite and various metal oxides.5 The gold peaks did not shift on the graphite and NiO(001) substrates. However, peak shifts of up to +0.4 eV were observed on SrTiO3, Al2Ox/NiAl(110), TiO2 and NiO(111) at sub-monolayer gold coverage. Importantly, on both SrTiO3 and graphite the full-width-half-maximum (FWHM) of the gold peak decreased slightly with increasing particle size. Lykhach et al. evaporated gold on Nb2O5/Nb, Nb2O5/W and NbO/Nb surfaces.35 They find shifts in the gold peak position of up to +0.7 eV, with the largest shifts observed for the smallest Au coverage. Lim et al. evaporated gold on SiO2/Si and observed shifts of the gold peak of up to +1 eV. Again, the largest shift corresponded to the smallest amount of gold evaporated. Exposure to atomic oxygen leads to the appearance of a second peak at about 86 eV, attributed to the formation of Au2O3.6
Deposition of size-selected clusters produced under gas-phase conditions have been known for over a decade. DiCenzo et al. measured Au 4f XPS data for gas phase, size-selected Aun clusters deposited on amorphous carbon. For cluster sizes of n = 5, 7, 27 and 33 they observed shifts of +0.7, +0.6, +0.5 and +0.5 eV, respectively, with peak FWHM being slightly broader for the smaller clusters.36 Cox et al. reported Au 4f7/2 XPS data for gas phase, size-selected positively charged clusters deposited on silica (natural oxide layer on Si(100)-wafers). They observed a shift of +0.8–1.0 eV for Aun (n = 1–7).37 More recently, Lim et al. have published a series of papers where mass-selected Au cluster anions up to 20 atoms were soft-landed onto SiO2/Si surfaces and the samples subsequently exposed to atomic oxygen.6,38–40 Although peak widths of 1.5 eV were observed, in addition to Au0 peaks, the clusters also exhibited features attributed to AuIII, which were shifted by +1.5 eV. Interestingly, an even–odd effect was observed, whereby the odd-numbered clusters were inert towards oxidation and exhibited very small AuIII peaks, particularly so for Au5, Au7 and Au13. These workers also deposited the same clusters on sputter-damaged HOPG. In this case, only Au8 showed any ability to oxidise after oxygen treatment. Small gold peak shifts of +0.1 eV to +0.2 eV were observed relative to bulk gold.
XPS studies have also been performed on chemically-synthesised, ligand protected gold species; from monoatomic Au compounds (e.g. Au(PPh3)(NO3)), to atomically precise Au clusters (e.g. Au8(PPh3)8(NO3)3), to even larger, less precise, nanoparticles spanning the size range from ca. 1.5 nm (“Au101”41) and above. Yuan et al. have deposited Au(PPh3)(NO3) on Ti(OH)4 and TiO2 showing a Au 4f7/2 of 85.5 eV in the first case and 84.8 eV in the latter case and attributed the difference in binding energy to different interactions of the cluster with the substrate.42 Chusuei et al. deposited [Au6(PPh3)6][BF4]2 clusters on TiO2 and observed Au 4f7/2 peaks with binding energy of 83.9 eV.43 Removal of the triphenylphosphine ligand groups via electron-stimulated desorption resulted in a shift of +0.4 eV. This was attributed to the removal of the ligands, which was corroborated by a reduction in size as observed by STM. Very early XPS studies by Battistoni et al. investigated Au1 Au4, Au8, Au9 and Au11 clusters stabilised by triphenylphosphine ligands.44,45 Samples deposited from alcohol solutions onto steel sample plates showed Au binding energies that were 0.7 to 1.7 eV higher than bulk gold, and which depended upon the number of ligands and the counter ions. By analysis of the different peak shapes they concluded that Au9 and Au11 clusters possess centred polyhedral geometries with a central metal atom involved only in metal–metal bonds. Van Attekum and co-workers similarly published an XPS analyses of chemically synthesised Au8, Au9 and Au11 stabilised by tri-arylphosphine ligands.46 Clusters were evaporated onto graphite substrates from solutions in dichloromethane. All clusters exhibited individual Au 4f7/2 peaks with widths of 1.5 eV. Only kinetic energy values have been tabulated and calibrated relative to the P peak, however, these could be subject to a chemical shift so the peak positions may not be reliable. Boyen et al. used XPS to investigate a series of gold nanoparticles made using polymers as stabilizing agents with metal core sizes ranging from 2.9 down to 0.8 nm deposited on silicon. Following removal of stabilizers by treatment first with oxygen and subsequently with hydrogen plasma, the authors found peak shifts of up to +0.5 eV at their smallest cluster size of 0.8 nm.47 The same authors found that phosphine stabilized 1.4 nm (similar size as Au101 (ref. 41)) nanoparticles after oxygen/hydrogen plasma treatment show an Au 4f7/2 peak at about 84.2 eV. Ono et al. reported for polymer-stabilised gold particles peak positions of about 85 eV for 1.5 nm gold and about 87 eV for oxidised 1.5 nm gold clusters on both TiO2 and SiO2 substrates following treatment at 150 K with oxygen plasma only.48 Upon heating, they observed very fast decomposition of gold oxide species (within 6 min at 350 K) for the nanoparticles supported on TiO2 which was in stark contrast to the case of SiO2-supported gold oxide species. More recently, Turner et al. found the Au 4f7/2 peak of 1.5 nm gold clusters deposited on BN at a binding energy of 85.1 eV.14
In summary, the smaller clusters tend to exhibit the highest binding energy shifts, with the binding energy decreasing as cluster size increases before finally converging to the bulk value at large cluster sizes.5,36,47,49,50 The FWHM also shows in many cases an increase with decreasing cluster size.5,35 The shift, however, also strongly depends on the substrate. For “inert” substrates such as HOPG and pure Si, only small shifts, <0.1 eV, are observed. SiO2 surfaces tend to shift the peaks by ∼0.2 eV to higher energy. Highly interacting surfaces such as TiO2, SrTiO3, Al2O3 and NiO(111) induce shifts up to 1.5 eV.
The goal of this paper is to identify changes in electronic structure for a series of chemically-synthesised, atomically-precise gold clusters that occur upon their deposition onto titania followed by selected treatments – washing and calcination under mild conditions. Using Synchrotron XPS we have monitored removal of ligands, changes attributed to partial agglomeration and interaction of the cluster core with the substrate.
Experimental
The clusters were synthesized according to well-established, previously reported methods. Each cluster has been recrystallised at least twice to achieve high purity. Furthermore, their identities have been confirmed by NMR, MS and X-ray crystallography (CCDC 907704 for Au11 and 907703 for Au8, Fig. 1 and ESI†). Specific details of the synthesis and purification via recrystallisation of the Aun clusters (n = 8, 9, 11, 101) is described in the ESI.† The following information regarding preparation of the support-immobilised clusters and treatments is presented below because it is essential to the discussion. | ||
| Fig. 1 Crystal structures of Au8 (left column), Au9 (centre column) and Au11 (right column) clusters showing the exact number of Au atoms per cluster core. Top row: space-filled models showing triphenylphospine ligands effectively protecting the metal cores buried underneath. Centre row: ball and stick model showing positions of all atoms except hydrogen, co-crystallisation solvent molecules and non-coordinated counter anions. Bottom row: geometries of the metal cores only. The atomic colour scheme is gold (Au), orange (P), green (Cl), and black (C). | ||
Deposition of clusters onto support
A solution of the cluster (20 mg) in methanol (30 mL) was added to the magnetically stirred suspension of acid washed Evonik P-25 (2 g) in methanol (50 mL) in a Schlenk tube. The suspension was left overnight and the solvent removed under vacuum taking care to avoid contamination of the Schlenk line with the resulting dry powder. The material obtained in this way was stored in a refrigerated Schlenk tube under N2 and in the absence of light.Evonik P-25 particles were chosen as substrates as this material is a very popular TiO2 support used by numerous researchers worldwide, which very recently was U.S. National Institute of Standards and Technology as a nanoscale reference material. Further, particles have a large number of surface defects which is assumed to support the adsorption on the surface.51 It has to be noted that Evonik P-25 is a mixture of 70% anatase and 30% rutile particles. The interaction between the gold clusters and anatase and rutile could differ52 resulting in two different types of surfaces after deposition of the gold clusters. Prior to deposition of the gold clusters, the titania particles were treated with sulphuric acid.18
Supported cluster treatment conditions
Solvent treatment (washing) was undertaken following a similar protocol described by Lopez-Sanchez et al.18 Typically, the magnetically stirred (1000 rpm) suspension of the supported cluster on TiO2 (ca. 500 mg) in toluene (50 mL) was heated to 100 °C for 2 hours. The solid was then recovered using centrifugation (10 min, 5000 rpm). The solid was then washed three times with toluene (50 mL) by forming a suspension using a Vortex agitator followed by centrifugation (10 min, 5000 rpm) to recover the solid, which was then dried under reduced pressure at about 10−2 mbar. The material obtained in this way was stored in a refrigerated Schlenk tube under N2 and in the absence of light.Toluene was chosen as a solvent in the washing procedure as it is frequently used in catalytic processes. Thus, washing treatment with toluene at elevated temperatures also mimics the effect of the induction period during the first two hours if untreated catalyst is used.
Heat treatment or calcination
We follow the protocol described by M. Turner et al.14 The supported cluster on TiO2 (ca. 500 mg) in a Schlenk tube was stirred (1000 rpm) and heated at 200 °C (oil bath) under vacuum for 2 hours. The sample was then removed from the oil bath and allowed to cool to room temperature under reduced pressure. The material obtained in this way was stored in a refrigerated Schlenk tube under N2 and in the absence of light.Sample preparation for XPS analysis
A suspension of the support-immobilised cluster was made up in dichloromethane at a concentration of ca. 1 mg mL−1. A 10 μL drop of each sample was deposited onto a clean 6 × 6 mm silicon (Si) wafer and dried in air. Each sample was then fixed by double-sided copper tape onto a gold-plate; up to 6 samples were affixed onto each plate for XPS analysis. On a representative number of samples, various spots were investigated. Between the investigated spots, no significant variation of the XPS signals was found. To confirm reproducibility of our results a completely new batch of the Au9 cluster was deposited on a new batch of acid washed TiO2 and the resulting material was heat treated as before. The XPS spectra for new “untreated” and “calcined” samples were in excellent agreement with earlier obtained data.Synchrotron beam line
Photoelectron spectra were recorded at the Soft X-ray Beamline at the Australian Synchrotron (AS) using a SPECS Phoibos 150 hemispherical electron analyser with the photon energy set to 650 eV. The beam was adjusted to yield an irradiation spot size of ∼600 × 600 μm, yielding an X-ray photon flux of approximately 1012 photons mm−2 s−1, conditions that have been shown not to induce thermal damage to samples of NaAuCl4.53 High resolution XPS spectra of C, O, Si, P, Ti and Au were recorded at a pass energy of 10 eV, yielding an instrumental resolution of 295 meV.54 Scans were repeated several times to ensure that no photon-induced changes occurred in the samples. The stability of the energy of the X-ray was monitored using a bulk gold reference.All XPS spectra were fitted in the following manner. A Shirley background was applied to remove the electron-scattering background and maintain the intrinsic line shape from the raw data.55,56 A pseudo-Voigt function composed of the sum of Gaussian (30%) and Lorentzian (70%) functions was used to fit all peaks and all peak positions were allowed to vary using nonlinear least-squares minimization.57 For the Au 4f doublet, the splitting was fixed at 3.67 eV while for the P 2p doublet a splitting of 0.84 eV was used.58 All spectra where fitted with the least number of peaks allowing a variation of the FWHM, although the FWHM of a single contributing species was kept constant. For example, in the case of a gold spectrum fitted with two sets of 4f7/2 and 4f5/2 doublets, the FWHM within a single doublet was kept constant but was allowed to differ between the doublets.
The position, the intensity and the FWHM of the peaks fitted to the Au 4f7/2 spectra including the errors are given in Table 1. As an example, the fitting of the Au9 spectra are shown in the ESI.†
| Untreated | Washed | Calcined | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Energy [eV] | Intensity rel. to untreated [%] | FWHM [eV] | Energy [eV] | Intensity rel. to untreated [%] | FWHM [eV] | Energy [eV] | Intensity rel. to untreated [%] | FWHM [eV] | |
| Au8 LBP | 85.1 ± 0.1 | 100 | 1.6 ± 0.2 | 84.8 ± 0.1 | 37 ± 10 | 1.5 ± 0.2 | 84.0 ± 0.1 | 7 ± 10 | 1.2 ± 0.2 |
| Au8 HBP | — | — | — | 85.6 ± 0.1 | 16 ± 20 | 1.3 ± 0.2 | 85.4 ± 0.1 | 19 ± 10 | 1.6 ± 0.1 |
| Au9 LBP | 85.1 ± 0.1 | 100 | 1.6 ± 0.2 | 84.8 ± 0.1 | 47 ± 10 | 1.7 ± 0.2 | 84.2 ± 0.1 | 12 ± 10 | 1.3 ± 0.2 |
| Au9 HBP | — | — | — | 85.9 ± 0.1 | 17 ± 10 | 1.2 ± 0.2 | 85.5 ± 0.1 | 33 ± 10 | 1.5 ± 0.1 |
| Au11 LBP | 84.7 ± 0.1 | 100 | 1.6 ± 0.2 | 85.0 ± 0.2 | 13 ± 20 | 1.9 ± 0.2 | 84.2 ± 0.1 | 15 ± 10 | 1.1 ± 0.2 |
| Au11 HBP | — | — | — | 86.1 ± 0.1 | 18 ± 10 | 1.3 ± 0.2 | 85.5 ± 0.1 | 40 ± 10 | 1.5 ± 0.1 |
| Au101 LBP | 83.9 ± 0.1 | 100 | 1.0 ± 0.2 | 83.9 ± 0.1 | 5 ± 5 | 1.0 ± 0.2 | 83.9 ± 0.1 | 28 ± 10 | 0.9 ± 0.2 |
| Au101 HBP | — | — | — | — | — | 84.8 ± 0.1 | 14 ± 10 | 2.3 ± 0.2 | |
Cross sections were calculated according to Yeh and Lindau using the photoionization cross section and the asymmetry parameter.59 The angle between the sample normal and the analyser was 10° which means that the angle between the incident horizontal linearly polarised synchrotron beam and the sample surface was 45°.
The preparation of reference samples and recording of XP spectra is described in the ESI.† The reference samples were prepared such that multilayers of untreated Aun clusters were deposited on UHV compatible double-sided, adhesive copper conducting tape (3M brand). Due to this sample preparation, the reference XP spectra are recorded from Aun clusters that are not in contact with a substrate, i.e. no cluster–substrate interaction occurs.
Results and discussion
The molecular clusters (n = 8, 9, 11) used in this study contain atomically precise, ideally monodisperse metal cores protected by phosphine ligands, while Au101 particles (also stabilised by phosphine ligands) have narrow particle size distribution centered around 1.5 nm. The crystal structures of Au11, Au9 and Au8 clusters are shown in Fig. 1, confirming that the cluster metal core contain exactly 8, 960 and 11 atoms of gold respectively, with all the gold atoms involved in forming the metal–metal bonded cluster core. The two new X-ray structures reported here (Au11 and Au8) show typical Au–Au bonds of 2.6437(9) to 3.1274(10) Å in Au11, and 2.5827(6) to 2.8924(7) Å in Au8. The gold cluster at the centre of the Au8 structure approximates the cyclohexane structure with one gold atom added above and below the ring. This gives a shape with a short axis of 2.5827(7) Å, and a distance across the ring that varies from 5.2533(5) to 5.4536(7) Å. The gold cluster in the Au11 structure is quite spherical with ten ligated gold atoms (either by triphenylphosphine or chloride) surrounding a central gold atom. The shortest distance across the cluster 4.695(2) Å and the longest is 5.385(2) Å. Further discussion of the XPS results will be carried out in the light of the atomic precision of the molecular cluster systems studied here.Spectra have been taken at the gold 4f, phosphorous 2p, silicon 2p, titanium 2p, carbon 1s and oxygen 1s regions for Aun clusters deposited on titania nanoparticles and from a gold reference sample. The majority of carbon on the samples is either carbon of the triphenylphosphine ligand of the gold cluster or adventitious hydrocarbons. In either case, we assigned the C 1s peak to 285 eV and used it for calibrating the peak positions of other elements. This is justified because adventitious hydrocarbons display a very constant C 1s peak position and are present in all samples exposed to air (and not cleaned by sputtering etc. prior to XPS measurements).61 The titanium signal measured is exclusively due to the titania nanoparticles and found at 459.0 ± 0.1 eV. The silicon signal is due to the silicon wafer used as substrate and found at 99.2 ± 0.1 eV (Si) and 103.1 ± 0.1 eV (SiO2). The oxygen signal is mostly due to titanium dioxide (530.3 ± 0.1 eV) and silicon dioxide (532.4 ± 0.1 eV). Gold signal due to the gold clusters is found between 83.7 and 86.1 eV. Phosphorous from the ligands is found at 131.8 ± 0.1 eV and 133.0 ± 0.2 eV. The Au and P signals are used for quantitative analysis. Using the energy calibration described above, the Au 4f7/2 peak of bulk gold is found at a binding energy of 84 eV. This value is used to determine the shift between the Au 4f7/2 signal of the gold clusters and the bulk gold binding energy. Most references for gold are found at a binding energy of 84 eV within a range of ±0.2 eV.62–65 The invariance of the titanium and silicon signals relative to the gold reference is less than 0.05 eV, indicating that there is no charging of the samples or instability in the X-ray energy. Small amounts of sulphur were found in the samples, which is due to the treatment of the titania support with H2SO4 prior to the deposition of clusters.
Our XPS analysis of the gold and phosphorous peaks requires these signals to be normalised. The intensity of the gold spectra, i.e. the integral over the peaks fitted to the spectra, has to be normalised as the silicon substrate was, in all cases, not fully covered with the titania particles, i.e. there is a variable loading with titania particles. Normalisation of the phosphorous signal is not required since only the ratio of the phosphorous to gold intensity is of interest. The normalisation of the gold intensity and the calculation of the phosphorous to gold ratio are described in detail in the ESI.†
Untreated samples
The Au 4f spectra for the Au8, Au9, Au11 and Au101 clusters are shown in Fig. 2 for the untreated (i.e. as deposited) clusters (A), the samples washed after deposition (B), and the calcined samples (C). In these spectra, it can be seen that the gold peaks shift towards higher binding energies with decreasing number of atoms in the metal cluster. All four spectra can be fitted with a single peak for each of the Au 4f7/2 and Au 4f5/2 components. The peak position and the FWHM are shown in Fig. 3A (as will be discussed below, these peaks are referred to as the gold low binding peak, Au-LBP). The positions of all peaks are at a higher binding energy than that of metallic gold except for Au101. The FWHM of Au8, Au9 and Au11 is the same within experimental error but are larger than that of Au101. Shifts in the peak position and an increase in the FWHM of the XPS signal of particles consisting of a small number of metal atoms has been attributed in a number of cases to finite size effects and depends on the element constituting the nano-sized object as well as the substrate on which they are deposited.5,46 In the case of gold, peak shifts up to +1 to +1.5 eV,4 +0.7 eV,35,36 +1 eV3 and +1.1 eV14,47 have been previously reported. In all cases, an increase of the FWHM relative to that of bulk gold signal has also been reported. Thus, we attribute the shift of the peak position to the decrease in size of the gold cluster. The peak positions relative to the bulk gold Au 4f7/2 found here are +1.1 eV for Au8 and Au9, +0.7 eV for Au11 and −0.1 eV for Au101. The position of Au 4f7/2 of the untreated samples is very close to the peak position of the reference spectra as shown in Fig. 3D. Thus, it can be concluded that the clusters as deposited are not affected by the titania substrate. The position of the Au 4f7/2 peaks for both the untreated gold clusters and the reference samples are in the range of the Au 4f7/2 position reported for nanosized gold. As an example, Battistoni et al.44 reported that for Au9(PPh3)8(NO3)3 deposited on steel plates, the Au 4f7/2 peak lies at 85.2 eV (using the same energy calibration as here). The position of the gold peak as a function of the diameter of the gold cluster is shown in Fig. 3E. The diameter d of the gold cluster is approximated with d ∼ N1/3 where N is the number of gold atoms in the cluster. For such small values of N, this approximation is only a rough estimation to the cluster size but even so, Fig. 3E has similarities to the 1/d relation found for gold nanoparticles by Boyen et al.47 Due to its large size Au101 has a binding energy the same as that of bulk gold (within experimental error). To the best of our knowledge, there is only one previous Au XPS study of a chemically-prepared, atomically precise gold cluster deposited on titania. Chusuei et al. observed a binding energy of 83.9 eV for Au6(PPh3)6(BF4)2 deposited onto a TiO2(110) surface,43 which is significantly lower than the values we observed. Chusuei et al. used a similar binding energy calibration to that used here.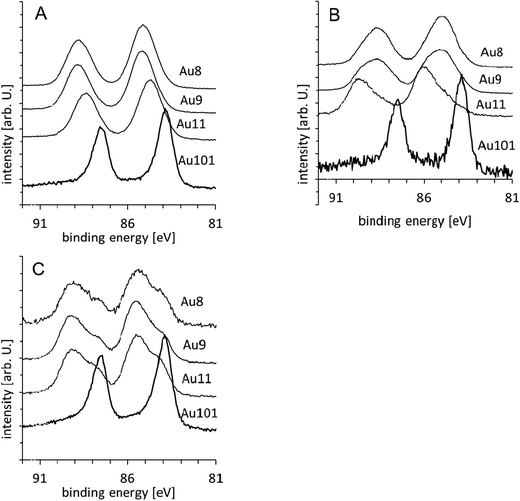 | ||
| Fig. 2 Au XPS spectra of Aun samples: (A) untreated, (B) washed and (C) calcined clusters. | ||
 | ||
| Fig. 3 Position of the Au-LBP and Au-HBP for the (A) untreated, (B) washed and (C) calcined clusters. Note that the Au-LBP peak of the washed Au101 cluster in (B) can be fitted with a single peak. In (D), the peak position of the gold clusters in the reference spectra are shown. In (E) the Au-LBP position and FWHM of the untreated clusters are shown as a function of the diameter, where the number of gold atoms to the power 1/3 is used to approximate the nanoparticle diameter. | ||
In Fig. 5A the phosphorous spectra of the untreated Au8, Au9 and Au11 cluster samples are shown. The phosphorous spectra from the Au101 samples were too weak to reliably separate the spectrum from the background and hence are not shown. The spectra are fitted with two peaks, one at 131.8 eV, which we will refer to as the phosphorous low binding energy peak (P-LBP), and another at 133.1 (±0.2) eV, which we will refer to as the phosphorous high binding energy peak (P-HBP). The position of the P-LBP is the same as that found in the reference samples and also the same as reported by Battistoni et al.44 We attribute the P-HBP to oxidised phosphorous which will be justified below. Fig. 6A and 5B show the ratios of the P-LBP and P-HBP, respectively, relative to the sum of the Au 4f7/2 signal. It can be seen that the P-LBP in the untreated samples is the dominant peak (i.e. intensity in Fig. 5A > Fig. 5B for all untreated clusters) and also that the sum of both ratios (Fig. 6C) is close to unity. In the spectrum of AuPPh3Cl (not shown here), only the P-LBP is observed with an intensity ratio between phosphorous and gold very close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Hence, the P-LBP in Fig. 5 is attributed to phosphorous of the ligands bound to gold. Higher binding energies of phosphorous (similar to that of P-HBP) are found for compounds with phosphorous bound to oxygen.66 Hence, it is concluded that in the untreated support-immobilised clusters there is about one ligand per gold atom and that the nature of the bond between the phosphorous of the ligand and the gold is very similar to that in AuPPh3Cl.
:1. Hence, the P-LBP in Fig. 5 is attributed to phosphorous of the ligands bound to gold. Higher binding energies of phosphorous (similar to that of P-HBP) are found for compounds with phosphorous bound to oxygen.66 Hence, it is concluded that in the untreated support-immobilised clusters there is about one ligand per gold atom and that the nature of the bond between the phosphorous of the ligand and the gold is very similar to that in AuPPh3Cl.
A molecular representation showing how the Au9(PPh3)8 cluster may be deposited onto the titania surface is shown in Fig. 7A, where all ligands remain attached to the cluster core and prevent its contact with the substrate.
Washed samples
In the Au 4f spectra of the washed samples (Fig. 2B) it is necessary to fit a second, higher energy, gold doublet for Au8, Au9 and Au11 (hereafter referred to as Au-HBP). Fig. 3B shows that the Au-LBP peak positions relative to bulk Au 4f7/2 are +0.8 eV for Au8 and Au9, +1.0 eV for Au11 and −0.1 eV for Au101 and only marginally different to those of the untreated samples. The FWHMs are the same within the experimental error as those for the Au-LBP in the untreated samples. The Au-HBP is found at +1.6 eV for Au8, at +1.9 eV for Au9, +2.1 eV for Au11. No Au-HBP peak is found for Au101. This binding energy is very similar to that of Au2O3 thin films which shows a shift in binding energy of 1.8 to 2.1 eV.6,31,32 Ono et al. have reported shifts in binding energies from about 85 eV to about 87 eV when exposing 1.5 nm sized gold clusters supported on TiO2 and on SiO2 to oxygen plasma (i.e. about 3 eV higher binding energy than bulk gold).48 It is worth noting that Au101 also has a size of about 1.5 nm. Lim et al. reported that gold peaks from gas-phase deposited Aun (n = 3–20) clusters deposited on SiO2 after exposure to atomic oxygen are found at about 86 eV with a shift of about 2 eV relative to the unexposed sample, which they attributed to the formation of gold oxide.6Fig. 4A shows the intensity of both the Au-LBP and Au-HBP. The intensities have been normalised as outlined above and then divided by the intensity of the untreated samples. In general, the total intensity of the peaks is lower than that of the untreated samples (i.e. ratio < 1). Furthermore, it can be seen that in the case of Au8 and Au9 the intensity of the Au-LBP is larger than that of the Au-HBP, while this is reversed for Au11.
 | ||
| Fig. 4 Ratio of the intensity of the Au-LBP and Au-HBP of (A) washed and (B) calcined clusters, relative to the intensity of the Au peak for the untreated cluster. | ||
As both the position and the FWHM of the Au-LBP are similar to that of the untreated clusters, it can be concluded that this fraction of the gold clusters is not significantly altered in its size and chemical state. We attribute the appearance of the Au-HBP to the formation of Au–O bonds involving the gold atoms of the cluster cores as the chemical shifts are significantly larger than that of the untreated clusters. We do not attribute it to fully oxidised gold clusters as the shift in peak position relative to the untreated samples is +0.8 to +1.1 eV and, thus, significantly less than the +2 eV shifts reported by others.38,48 Whether the shift is due to the formation of partially oxidised gold clusters or due to a strong interaction of the flat (2D), “raft-like” gold cluster core with the TiO2 support cannot be distinguished. Earlier reports suggest that XPS spectra of the ultra-small “raft-like” metal clusters can be strongly affected due to metal–support interactions.67 However, it has to be noted that previously oxidation of gold clusters has only been achieved by using a strong oxidising agent.6,31,32 In contrast, we have heated the samples to 100 °C in toluene which is not a strong oxidising environment for the clusters. Hence, the oxidation of the gold with oxygen due to the traces of air dissolved in the liquid phase is less likely than the formation of Au–O bonds with oxygen of the titania surface. For the latter, some of the ligands of the gold cluster have to be removed to allow for the formation of Au–O bonds. The removal of some or all ligands is considered necessary in the activation of the clusters for catalysis but68 to the best of our knowledge this report provides the first ever evidence of the formation of Au–O bonds between metal cores of chemically-synthesised atomically-precise clusters and oxide support material due to the removal of ligands induced by mild treatment. The removal of ligands also assists in shifts to higher binding energy as ligands are electron donors. In principle, the oxygen signal could be used to further prove this assertion. Unfortunately, the oxygen peaks are dominated by contributions from SiO2 and TiO2 and also, the binding energy of oxygen in TiO2 and gold oxide is very similar and thus would not provide a distinction between these two species.
Alternatively, it could be argued that the Au-HBP is due to the formation of bulk gold oxide after agglomeration of the gold clusters. We consider this option as unlikely for the same reason as before – that the applied treatment is not a strong enough oxidiser to form an oxide layer on bulk gold. Therefore, the overall reduction in gold intensity has to be interpreted as removal of clusters from the surface through the washing procedure.
The Au101 clusters show a peak at the same position and with the same FWHM as the untreated sample but with a very significant reduction in intensity. Therefore, it seems that the washing process removes Au101 clusters from the sample but does not change the chemical state of the Au101 cluster core. It has to be noted that Au101 clusters are easiest to remove by washing.
In Fig. 5B, the P spectra of the washed Au8, Au9 and Au11 cluster samples are shown. The spectra show peaks with the same position and FWHM as the untreated samples. Fig. 6A and B show the ratio of the P-LBP and P-HBP, respectively, relative to the sum of the Au 4f7/2 signal. It can be seen that in the washed samples the P-LBP is the dominant form, which is similar to the spectra of the untreated samples, as seen in Fig. 6C. The sum of both ratios is close to unity for the Au8 and Au9 clusters but is around 1.3 for the Au11 cluster. In the latter case, however, we ascribe this to the relatively large uncertainty in peak area.
 | ||
| Fig. 5 P XPS spectra of (A) untreated, (B) washed and (C) calcined clusters. The spectrum for Au101 is not shown as the P signal was too weak to be used for reliable subtraction of the background. | ||
 | ||
| Fig. 6 Ratio of the intensity of (A) the P-LBP, (B) the P-HBP, and (C) the P-HBP + P-LBP, to the sum of the intensity of the respective Au peaks (i.e. both Au-LBP and Au-HBP). | ||
It could be argued that for the formation of Au–O bonds some ligands have to be removed and hence the P/Au intensity should decrease but this is not observed. However, the uncertainty of the phosphorous signal is too large to conclude that this is a contradiction.
A molecular representation showing how the Au9(PPh3)8 cluster may interact with the titania surface following washing is shown in Fig. 7B. One of the ligands is removed enabling the cluster core to make contact with the substrate and form Au–O bonds, which are observed as Au-HPB. It should be emphasised that Fig. 7B is for illustration only and that we do not have evidence for the exact structure of the cluster core when it is in contact with the substrate.
 | ||
| Fig. 7 Schematic illustration of the changes of the clusters due to washing and calcination. In (A) the cluster is shown after deposition (Au9(PPh3)8). The ligands are attached to the cluster core and the core is not in contact with the substrate. In (B), one of the ligands is removed (i.e. Au9(PPh3)7) and the core is in contact with the substrate. (Note: whilst these figures are based upon the crystal structures of the Au9 cluster and anatase (101) surface separately, the relative position and orientation of the cluster–surface interaction does not involve any geometric or energy minimisation process. They should therefore be regarded as crude representations of a possible interaction that is consistent with the XPS data. It should also be noted that the counter ion is not shown. Furthermore, it must be emphasised that the exact number of ligands removed is unknown and may be more than shown here.) | ||
Calcined samples
The Au 4f spectra of the calcined samples are shown in Fig. 2C. Again, it is clear that two doublets are required to fit the experimental spectra. Fig. 3C shows that the positions of the Au-LBP found here relative to bulk Au 4f7/2 are 0.0 eV for Au8, +0.2 eV for Au9 and Au11 and −0.1 eV for Au101 and thus are only marginally different to that of bulk gold. The FWHM for these peaks is ca. 1.2 eV, which is smaller than those of the untreated and the washed samples. Based on the trend of the untreated samples (Fig. 3A and C) we attribute the decrease in binding energy of the Au-LBP after calcination to agglomeration of the clusters. The change in the gold binding energy allows us to conclude that the size of the agglomerated clusters is only marginally smaller than that of untreated Au101, which is also reflected in the FWHM of the peaks as they are only little larger than that of Au101. The Au-HBP relative to bulk Au 4f7/2 is found at +1.4 eV for Au8, +1.5 eV for Au9 and Au11 and +0.8 eV for Au101. The FWHM of these peaks is 1.5 to 1.6 eV and is similar to that of ultra-small, untreated, support-immobilised clusters. In the same way as for the washed samples, we attribute the Au-HBP to the formation of Au–O bonds involving the gold atoms of the cluster cores as the chemical shift is similar to, but slightly (0.2 to 0.6 eV) smaller than that of the washed samples. In this case, the peak positions are too low in binding energy for assigning these peaks to either fully oxidised gold clusters or oxidised bulk gold. Here again, we cannot pinpoint whether the shift is due to the formation of partially oxidised gold clusters or due to a strong interaction of the flat (2D), “raft-like” gold cluster cores with the TiO2 support. Heating at 200 °C in low vacuum is a somewhat harsher environment than the washing procedure but still cannot be considered as a strong oxidising treatment comparable to oxygen plasma. Also, gold oxide has been found to be unstable at the temperatures applied here. Hence, also in this case the oxidation of the gold with gas phase oxygen is unlikely and the formation of Au–O bonds with oxygen of the titania surface has to be considered as being much more likely. The shifts are slightly reduced relative to the washed samples. The reason could be some degree of agglomeration of the clusters that have formed Au–O bonds under higher temperature conditions.The total intensity of gold is significantly reduced compared to the untreated sample as can be seen in Fig. 4B, with the Au-HBP being more intense than the Au-LBP. Different to washing, calcination does not remove clusters from the support and hence the amount of gold in the sample must be the same as for the untreated samples. Thus, the decrease in gold intensity can only be attributed to agglomeration of the gold cores of the clusters. An increase in gold particle size means that the signal of the gold from the side of the cluster closer to the substrate is attenuated due to the limited mean free path of the photoelectrons. As the amount of gold on the sample is constant, the total gold intensity decreases. This interpretation is supported by the fact that the position of the Au-LBP is close to 84 eV.
Fig. 5 shows the phosphorous spectra of the calcined Au8, Au9 and Au11 cluster samples. The spectra clearly illustrate a shift in the peak position compared to the spectra of the untreated samples from the P-LBP to the P-HBP position. A similar shift of the phosphorous peak was reported earlier after calcination of Au(PPh3)(NO3) on TiO2.42 Based on XPS results and 31P-NMR, Yuan et al., assigned the change of their sample to either the formation of species where phosphine is bound to a larger metal particle, i.e. the formation of gold clusters surrounded by triphenylphosphine ligands, or the oxidation of phosphorous. Based on our results presented here, we can rule out the first possibility as the untreated Aun clusters show a lower binding energy of the phosphorous. Hence, we ascribe the P-HBP to oxidised phosphorous species which is also supported by the peak position of phosphorous in phosphorous-doped titania.69 Noteworthy, the ratio of the P-LBP and the P-HBP to the sum of the gold 4f signal (see Fig. 6B) has almost entirely shifted to the P-HBP peak for the Au8 and Au11 clusters and to some extent for the Au9 cluster. The sum of both ratios is larger than unity for the Au8 and Au11 clusters. This finding can be explained by gold clusters agglomerating into larger particles during calcination, consistent with the Au-LBP signal shifting to lower binding energy and the overall decrease in total gold peak intensity. The extraneous ligand P moves elsewhere onto the surface and becomes oxidised or bonded to the titania surface resulting in an increase in the P-HBP signal (see Fig. 6B). The formation of larger gold particles should reduce the gold signal due to the limited electron mean free path leading to an increase in the total P:Au ratio (see Fig. 6C) and also to an increase in the P-HBP signal (see Fig. 6B). The amount of “ligand-like” phosphine species (P-LBP) is reduced by ca. 50% due to the maintained presence of a fraction of the non-agglomerated clusters even in calcined samples (see Fig. 6A). This interpretation is supported for Au8 and Au11 by the changes in the phosphorous signal shown in Fig. 6. To a lesser degree the interpretation is also supported for Au9. However, the removal of the ligands from the cluster and their oxidation seems to be less efficient for Au9 compared to Au8 and Au11, as the P-LBP is stronger in the case of Au9 compared to Au8 and Au11. In summary, it can be concluded that the phosphorous of the ligands oxidises during calcination and is most likely removed from the clusters but remains in an oxidised form on the surface. The changes of the clusters deposited on the titania particles due to the calcination procedure applied are illustrated in Fig. 7. In terms of the molecular representation in Fig. 7B, although calcining of the sample does lead to removal of ligands, it is also observed that agglomeration of the gold core occurs.
Conclusions
We have investigated chemically synthesised, atomically precise gold clusters deposited on titania nanoparticles using XPS. Subsequent to their immobilisation on the titania support, the gold clusters have been treated with washing and calcination to remove the phosphine ligands from the gold cluster core. XPS has been used as a tool to determine changes in the chemical nature, composition, and to estimate changes in the size of the gold clusters. Upon treatment three changes of the clusters have been found: firstly, removal of the phosphine ligands from the Au clusters and interaction of the cluster core with the substrate, secondly, partial agglomeration of the Au clusters and thirdly, oxidation of the phosphine ligands. The removal of the ligands is most likely an essential step to make the clusters catalytically active and their interaction with the substrate will affect their catalytic activity since it influences their charge state. Agglomeration of the clusters will also impact the catalytic properties of the deposited clusters. It is known that small clusters show size specific catalytic activity8,9 which is lost when they agglomerate. However, only partial agglomeration is found here and a portion of the clusters are not affected. As the ligands are at least partially removed, the non-agglomerated clusters could be involved in catalytic reactions. The interaction with the substrate could also affect the catalytic activity.We find that upon depositions, untreated clusters remain virtually unchanged and can be described as ultra-small clusters with decreasing size from Au101 to Au8, as indicated by positions and widths of the gold and phosphorus peaks in the XPS spectra. The peak positions in the gold XPS spectra shift systematically in agreement with the size of the clusters determined by X-ray diffraction immediately after their synthesis.
Washing of the support-immobilised cluster with toluene at 100 °C leads to the removal of a fraction of the clusters from the titania surface. Of those clusters left on the titania surface some remain virtually unchanged. The other fraction shows formation of Au–O bonds, most likely to the oxygen of the titania surface, correlating with at least partial removal of ligands. No significant agglomeration of the cluster cores is evident from the observed XPS spectra for this fraction.
Calcination of the support-immobilised clusters has two effects. First, the ultra-small clusters aggregate to form slightly larger gold particles that are still protected by phosphine ligands. The average size of the aggregated clusters can be estimated to be marginally smaller than that of untreated Au101. Second, a fraction of gold clusters forms Au–O bonds, again most likely to the oxygen of the titania surface, which coincides with the loss of phosphine ligands and formation of oxidised phosphorous species. Of this fraction of clusters, at least a portion does not show agglomeration.
Acknowledgements
This work was supported by a grant of the Australian Synchrotron (AS112/SXR/3576), the MacDiarmid Institute and the College of Science at the Canterbury University. The XPS measurements were undertaken on the soft X-ray beamline at the Australian Synchrotron, Victoria, Australia.References
- P. L. J. Gunter, J. W. Niemantsverdriet, F. H. Ribeiro and G. A. Somorjai, Catal. Rev.: Sci. Eng., 1997, 39, 77–168 CAS.
- F. H. Ribeiro, G. A. Somorjai, G. Wedler, B. C. Gates, C. T. Campbell, C. Xu, D. W. Goodman, A. Zecchina, D. Scarano, S. Bordiga, A. Cimino, F. S. Stone, Y. Iwasawa, M. A. Barteau, J. M. Vohs and J. P. Vigneron, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH, 2008, pp. 771–908 Search PubMed.
- M. S. Chen and D. W. Goodman, Chem. Soc. Rev., 2008, 37, 1860–1870 RSC.
- M. Büttner and P. Oelhafen, Surf. Sci., 2006, 600, 1170–1177 CrossRef.
- Y. Kitsudo, A. Iwamoto, H. Matsumoto, K. Mitsuhara, T. Nishimura, M. Takizawa, T. Akita, Y. Maeda and Y. Kido, Surf. Sci., 2009, 603, 2108–2114 CrossRef CAS.
- D.-C. Lim, C.-C. Hwang, G. Gantefor and Y. D. Kim, Phys. Chem. Chem. Phys., 2010, 12, 15172–15180 RSC.
- U. Heiz and W.-D. Schneider, Crit. Rev. Solid State Mater. Sci., 2001, 26, 251–290 CrossRef CAS.
- S. Kunz, K. Hartl, M. Nesselberger, F. F. Schweinberger, G. Kwon, M. Hanzlik, K. J. J. Mayrhofer, U. Heiz and M. Arenz, Phys. Chem. Chem. Phys., 2010, 12, 10288–10291 RSC.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS.
- R. Schlögl, M. Che, O. Clause, C. Marcilly, C. Louis, H. Knöinger, E. Teglauer, W. Keim, B. Drieben-Hölscher, J. W. Geus, A. J. van Dillen and J. Barbier, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH, 2008, pp. 138–264 Search PubMed.
- R. M. Crooks, M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- A. B. Hungria, R. Raja, R. D. Adams, B. Captain, J. M. Thomas, P. A. Midgley, V. Golovko and B. F. G. Johnson, Angew. Chem., Int. Ed., 2006, 45, 4782–4785 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS.
- R. Raja, V. B. Golovko, J. M. Thomas, A. Berenguer-Murcia, W. Z. Zhou, S. H. Xie and B. F. G. Johnson, Chem. Commun., 2005, 2026–2028 RSC.
- Y. M. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Chem. Commun., 2010, 46, 550–552 RSC.
- Y. M. Liu, H. Tsunoyama, T. Akita, S. H. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS.
- Y. Zhu, H. Qian and R. Jin, J. Mater. Chem., 2011, 21, 6793–6799 RSC.
- C. C. Chusuei, X. Lai, K. A. Davis, E. K. Bowers, J. P. Fackler and D. W. Goodman, Langmuir, 2001, 17, 4113–4117 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- R. Meyer, C. Lemire, S. Shaikhutdinov and H. Freund, Gold Bull., 2004, 37, 72–124 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS.
- N. Yi, R. Si, H. Saltsburg and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2010, 95, 87–92 CrossRef CAS.
- N. Yi, R. Si, H. Saltsburg and M. Flytzani-Stephanopoulos, Energy Environ. Sci., 2010, 3, 831–837 CAS.
- W. Huang, H.-J. Zhai and L.-S. Wang, J. Am. Chem.Soc., 2010, 132, 4344–4351 CrossRef CAS.
- Z. Jiang, W. Zhang, L. Jin, X. Yang, F. Xu, J. Zhu and W. Huang, J. Phys. Chem. C, 2007, 111, 12434–12439 CAS.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS.
- S. Porsgaard, P. Jiang, F. Borondics, S. Wendt, Z. Liu, H. Bluhm, F. Besenbacher and M. Salmeron, Angew. Chem., Int. Ed., 2011, 50, 2266–2269 CAS.
- K. Juodkazis, J. Juodkazytė, V. Jasulaitienė, A. Lukinskas and B. Šebeka, Electrochem. Commun., 2000, 2, 503–507 CrossRef CAS.
- B. Koslowski, H. G. Boyen, C. Wilderotter, G. Kästle, P. Ziemann, R. Wahrenberg and P. Oelhafen, Surf. Sci., 2001, 475, 1–10 CrossRef CAS.
- V. Borman, M. Pushkin, V. Tronin and V. Troyan, J. Exp. Theor. Phys., 2010, 110, 1005–1025 CrossRef CAS.
- S. Zafeiratos and S. Kennou, Surf. Sci., 1999, 443, 238–244 CrossRef CAS.
- Y. Lykhach, J. Plšek, I. Spirovová and Z. Bastl, Collect. Czech. Chem. Commun., 2003, 68, 1791–1804 CrossRef CAS.
- S. B. DiCenzo, S. D. Berry and E. H. Hartford, Jr., Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 8465–8468 CrossRef.
- D. M. Cox, W. Eberhardt, P. Fayet, Z. Fu, B. Kessler, R. D. Sherwood, D. Sondericker and A. Kaldor, Z. Phys. D: At., Mol. Clusters, 1991, 20, 385–386 CrossRef CAS.
- D. C. Lim, R. Dietsche, G. Ganteför and Y. D. Kim, Chem. Phys. Lett., 2008, 457, 391–395 CrossRef CAS.
- D. C. Lim, R. Dietsche, G. Ganteför and Y. D. Kim, Chem. Phys., 2009, 359, 161–165 CrossRef CAS.
- D. C. Lim, R. Dietsche, G. Ganteför and Y. D. Kim, Appl. Surf. Sci., 2009, 256, 1148–1151 CrossRef CAS.
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890–12891 CrossRef CAS.
- Y. Yuan, K. Asakura, A. P. Kozlova, H. Wan, K. Tsai and Y. Iwasawa, Catal. Today, 1998, 44, 333–342 CrossRef CAS.
- C. C. Chusuei, X. Lai, K. A. Davis, E. K. Bowers, J. P. Fackler and D. W. Goodman, Langmuir, 2001, 17, 4113–4117 CrossRef CAS.
- C. Battistoni, G. Mattogno, R. Zanoni and L. Naldini, J. Electron Spectrosc. Relat. Phenom., 1982, 28, 23–31 CrossRef CAS.
- C. Battistoni, G. Mattogno, F. Cariati, L. Naldini and A. Sgamellotti, Inorg. Chim. Acta, 1977, 24, 207–210 CrossRef CAS.
- P. M. T. M. Van Attekum, J. W. A. Van der Velden and J. M. Trooster, Inorg. Chem., 1980, 19, 701–704 CrossRef CAS.
- H. G. Boyen, A. Ethirajan, G. Kästle, F. Weigl, P. Ziemann, G. Schmid, M. G. Garnier, M. Büttner and P. Oelhafen, Phys. Rev. Lett., 2005, 94, 016804 CrossRef.
- L. K. Ono and B. Roldan Cuenya, J. Phys. Chem. C, 2008, 112, 4676–4686 CAS.
- C. N. R. Rao, V. Vijayakrishnan, H. N. Aiyer, G. U. Kulkarni and G. N. Subbanna, J. Phys. Chem., 1993, 97, 11157–11160 CrossRef CAS.
- A. K. Santra and D. W. Goodman, J. Phys.: Condens. Matter, 2003, 15, R31 CrossRef CAS.
- L. Ellis-Gibbings, V. Johansson, R. B. Walsh, L. Kloo, J. S. Quinton and G. G. Andersson, Langmuir, 2012, 28, 9431–9439 CrossRef CAS.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, Nat. Chem., 2011, 3, 489–492 CAS.
- Y.-Y. Fong, B. R. Visser, J. R. Gascooke, B. C. C. Cowie, L. Thomsen, G. F. Metha, M. A. Buntine and H. H. Harris, Langmuir, 2011, 27, 8099–8104 CrossRef CAS.
- B. C. C. Cowie, A. Tadich and L. Thomsen, AIP Conf. Proc., 2010, 1234, 307–310 CrossRef.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- J. Végh, J. Electron Spectrosc. Relat. Phenom., 2006, 151, 159–164 CrossRef.
- H. P. Hughes and J. A. Scarfe, J. Phys.: Condens. Matter, 1996, 8, 1421 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics, Inc., Eden Prairie, 1995 Search PubMed.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- F. Wen, U. Englert, B. Gutrath and U. Simon, Eur. J. Inorg. Chem., 2008, 106–111 CrossRef CAS.
- D. Briggs and M. P. Seah, Practical surface analysis, Wiley, Chichester, New York, Aarau, 1990 Search PubMed.
- K. Asami, J. Electron Spectrosc. Relat. Phenom., 1976, 9, 469–478 CrossRef CAS.
- K. Dückers and H. P. Bonzel, Surf. Sci., 1989, 213, 25–48 CrossRef.
- T. Darrah Thomas and P. Weightman, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 5406–5413 CrossRef.
- M. P. Seah, G. C. Smith and M. T. Anthony, Surf. Interface Anal., 1990, 15, 293–308 CrossRef CAS.
- C. A. Strydom and H. J. Strydom, Inorg. Chim. Acta, 1989, 159, 191–195 CrossRef CAS.
- A. Y. Stakheev and L. M. Kustov, Appl. Catal., A, 1999, 188, 3–35 CrossRef CAS.
- A. Kulkarni, R. J. Lobo-Lapidus and B. C. Gates, Chem. Commun., 2010, 46, 5997–6015 RSC.
- L. Lin, R. Y. Zheng, J. L. Xie, Y. X. Zhu and Y. C. Xie, Appl. Catal., B, 2007, 76, 196–202 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 907703 and 907704. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cp44005b |
| This journal is © the Owner Societies 2013 |