 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Raman microspectroscopy and vibrational sum frequency generation spectroscopy as probes of the bulk and surface compositions of size-resolved sea spray aerosol particles†
Andrew P.
Ault‡
a,
Defeng
Zhao‡§
b,
Carlena J.
Ebben
c,
Michael J.
Tauber
b,
Franz M.
Geiger
*c,
Kimberly A.
Prather
*bd and
Vicki H.
Grassian
*a
aDepartment of Chemistry, University of Iowa, Iowa City, Iowa 52242, USA. E-mail: vicki-grassian@uiowa.edu
bDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, USA. E-mail: kprather@ucsd.edu
cDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA. E-mail: geigerf@chem.northwestern.edu
dScripps Institution of Oceanography, University of California, San Diego, La Jolla, California 92093, USA
First published on 19th February 2013
Abstract
Sea spray aerosol (SSA) represents one of the largest aerosol components in our atmosphere. SSA plays a major role in influencing climate; however the overall impacts remain poorly understood due to the overall chemical complexity. SSA is comprised of a mixture of inorganic and organic components in varying proportions that change as a function of particle size and seawater composition. In this study, nascent SSA particles were produced using breaking waves, resulting in compositions and sizes representative of the open ocean. The composition of individual SSA particles ranging in size from ca. 0.15 to 10 μm is measured using Raman microspectroscopy, while the interfacial composition of collections of size-resolved particles is probed by sum frequency generation (SFG). Raman spectra of single particles have bands in the 980 to 1030 cm−1 region associated with the symmetric stretch of the sulfate anion, the 2800 to 3000 cm−1 region associated with carbon–hydrogen stretches, and from 3200–3700 cm−1 associated with the oxygen–hydrogen stretches of water. The relative intensities of these features showed a strong dependence on particle size. In particular, submicrometer particles exhibited a larger amount of organic matter compared to supermicrometer particles. However, for external surfaces of homogeneous SSA particles (i.e. particles without a solid inclusion), and also the interfaces of mixed-phase particles, there was a strong SFG response in the aliphatic C–H stretching region for both sub- and supermicrometer particles. This finding suggests that organic material present in supermicrometer particles primarily resides at the interface. The presence of methylene contributions in the SFG spectra indicated disordered alkyl chains, in contrast to what one might expect for a surfactant layer on a sea salt particle. Changes in peak frequencies and relative intensities in the C–H stretching region are seen for some particles after the addition of bacteria, phytoplankton, and growth medium to the seawater. This study provides new insights into the bulk and surface composition of SSA particles and represents a step forward in our understanding of this globally abundant aerosol. It also provides insights into the development of model systems for SSA that may more accurately represent the organic layer at the surface.
1. Introduction
Oceans cover 71% of the earth's surface, and the flux of sea spray aerosol (SSA) is the largest source of aerosol particles in the atmosphere.1–3 SSA particles influence the earth's climate by absorption and scattering of solar radiation and through their propensity to act as cloud condensation nuclei (CCN) and ice nuclei (IN).4–6 Climate-relevant physicochemical properties of aerosols, including CCN/IN activity and optical properties, depend on particle size and chemical composition, which, in turn, are controlled by chemical and physical interactions that occur at the interface between the particle and gas phase, a region that is notoriously difficult to access experimentally.SSA particles have been historically regarded as being mainly composed of sea salt, i.e. NaCl, as well as other inorganic salts.2,7 However, organic matter in the ocean, which can originate from exopolymeric secretions or marine organisms, includes complex mixtures of polysaccharides, lipids, and proteins in dissolved and particulate forms.8–12 It has long been known that SSA particles contain organic carbon species.13,14 Only recently have measurements provided detailed insight regarding the organic species present in SSA.3,4,15 From H-NMR analysis, water-soluble organic material in SSA was reported to be comprised of aliphatic moieties along with oxidized functional groups, including hydroxylated moieties.15,16 The water-insoluble portion of the major fraction of primary organic matter in SSA particles has recently been shown to be structurally similar to lipids, which are mainly aliphatic chains with terminal methyl groups.17 Measurements investigating the size-resolved chemistry of SSA particles from pristine marine conditions have shown that organic species are prevalent in the submicrometer size range, especially in particles smaller than 500 nm, while sea salt was found to dominate the chemical composition in the supermicrometer size range.4,15,16 Not surprisingly, it has also been reported that during periods of high biological activity, the average mass fraction of organic species increases for particles of all sizes.4 A similar increase in the mass fraction of organic matter was observed for particles generated from artificial seawater enriched with marine microorganism exudates.18
In general, the composition of aerosols can be determined by measurement of many particles15–17 or by analysis of single particles.19,20 The analysis of single particles can reveal not only their chemical constituents, but also their phase and mixing state (how species are mixed in different populations of particles).5,21–23 Experimental probes of single aerosol particles include scanning electron microscopy (SEM) or transmission electron microscopy (TEM)21,22 coupled to energy-dispersive X-ray analysis (EDX),24–26 scanning transmission X-ray microscopy with near-edge X-ray absorption fine structure microspectroscopy (STXM-NEXAFS),27 attenuated total reflection Fourier transform infrared imaging (ATR-FTIR),28 particle analysis by laser mass spectrometry (PALMS),5 and aerosol time of flight mass spectrometry (ATOFMS).23,29 Raman microspectroscopy provides a great deal of information regarding the chemical composition and speciation of individual SSA particles. Vibrational sum frequency generation (SFG), although not a single-particle approach, provides surface-specific insights that are complementary to Raman microspectroscopy. These spectroscopic techniques operate at ambient relative humidity and atmospheric pressure, avoiding artifacts introduced by exposure to vacuum during the measurement, such as loss of semi-volatile organic species. Raman microspectroscopy has been used for investigating heterogeneous chemistry,30 phase separation,31 hygroscopicity,32 and ice nucleation activity of model aerosol particles.33–38 However, only a few studies exist of Raman measurements on particles collected in the field,30,39–43 and Raman microspectroscopy studies on nascent SSA have been limited.40,42 Vibrational SFG has been recently used to identify C–H oscillators for a variety of environmentally relevant particle samples, including size-resolved secondary organic aerosol particles from tropical and boreal forests.44
In this study, data are presented and analyzed for size-segregated SSA particles generated from seawater using real breaking waves in the wave flume of the Scripps Institution of Oceanography (SIO) Hydraulics Laboratory before and after the addition of bacteria and phytoplankton. Raman microspectroscopy and vibrational SFG spectroscopy reveal the presence of organic matter in the sub- and supermicrometer sized particles. As discussed here, insights at the molecular level, including identification of functional groups are critical to assessing the impact of SSA particles on atmospheric chemistry and climate.
2. Experimental section
2.1 Aerosol generation and collection
All SSA particle samples were collected in November 2011 during an intensive campaign of the Center for Aerosol Impacts on Climate and the Environment (CAICE), the details of which are provided elsewhere.45,46 Briefly, the flume was filled with fresh seawater pumped from the end of the SIO pier that was coarsely filtered by two 3 × 7 m beds comprised of 30 cm of No. 12 crystal sand (∼1.7 mm diameter), 20 cm of pea gravel, and 45 cm of rock. The filters removed large biological and organic material and reduced bacteria concentrations by 75%.45 Waves were produced by a hydraulic paddle at the end of the wave flume and broke over an artificial beach sloped at a 30° angle relative to the bottom of the flume. Prior work has established that the physics of the breaking waves and the distribution of bubble sizes are comparable to those of waves in the open ocean.45,47 This method produced consistent particle concentrations of 100–150 particles per cm3.45 Given the low concentrations, a small contribution from background aerosol particles was unavoidable. However, the background count was kept to the lowest possible percentage (7–18%, or 10–18 particles per cm3) via an air filtration system and flushing flow rate of approximately 570 L min−1.45 For Raman analysis, an 8-stage micro-orifice uniform deposition impactor (MOUDI) (MSP Corp., Model 100) was used to collect size-resolved particles on a variety of substrates, including Ti (ESPI Metals, Inc., Model Knd1716) or Al (MSP Corp., Part 0100-96-0573A-X) foil and fused quartz windows (Ted Pella, Inc., Part 26016). For SFG measurements, particles were collected using a second MOUDI (MSP Corp., Model 100), as well as a PM1 sampler. Particles were collected on Teflon filters (Pall Life Sciences, Part 28139-125), which were stored at −12° C until just prior to analysis.482.2 Particle characterization
Particles in the supermicrometer and submicrometer size ranges were analyzed using two different micro-Raman spectrometers. The first (inVia, Renishaw) was equipped with Leica 50× and Olympus 100× objectives having numerical apertures of 0.5 and 0.8, respectively. A diode-pumped solid-state (DPSS) laser operating at 532 nm was focused on individual particles with incident power ∼2.5 mW. Backscattered light was collected at 180° with the same objective and passed through a notch filter to reject Rayleigh scattered light. The signal was dispersed in a single spectrograph equipped with an 1800 grooves per mm grating, and detected with a CCD. The second Raman spectrometer was a Nicolet Almega XR Raman spectrometer with a 532 nm DPSS laser and a 100× objective with a 0.9 numerical aperture. Raman spectra in the range of 500 to 4000 cm−1 were obtained with typical exposure times in the range of 10–60 s and a spectral resolution of 2–3 cm−1. All Raman spectra were analyzed following multi-line baseline correction. Both spectrometers were used for this study. The first spectrometer was used in San Diego during the CAICE intensive experiment for initial measurements of particle composition. The second spectrometer, located at the University of Iowa, was used to optimize instrument parameters for spectral quality, provide measurements of many more particles, and to further analyze particles from specific time periods during the course of the intensive experiment. Nearly all of the data reported herein were collected at the University of Iowa but were compared to the data collected during the CAICE intensive to ensure samples had not changed.Selected particles were characterized using an SEM (Phillips XL30 ESEM) equipped with Oxford EDX attachment and Inca software to obtain the particle images and elemental composition. A subset of these particles, whose positions on the substrate were carefully labeled, was first characterized using Raman microspectroscopy and then characterized using SEM-EDX to obtain the chemical speciation and elemental composition from the same particle. An accelerating voltage of 5 kV was used to show the light elements, especially carbon, while reducing the damage of the electron beam to the particle, and a higher accelerating voltage of 7.5 kV was used to obtain a better signal for the heavier elements, including K and Ca.
The details of the SFG setup are published in the literature.49,50 Briefly, a regeneratively amplified Ti:Sapphire laser system (Spectra Physics Spitfire Pro) operating at a 1 kHz repetition rate, which produced ∼120 fs pulses of 800 nm light was used. The beam passed through a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 beam splitter, and half was used to pump an optical parametric amplifier (Spectra Physics OPA 800C) which generated broadband IR pulses centered at ∼3.4 μm. SFG spectra were collected using a hybrid scanning/broadband method pioneered by Esenturk and Walker.51 Using this method, the IR light field was incident on the sample at several center wavelengths so that the full frequency range of interest was subjected to approximately the same incident IR power. This method allowed us to cover the CH stretching frequency region between 2700 and 3200 cm−1. To prevent optical damage to the samples, the incident pulse energies were limited to 1 μJ for the 800 nm beam and ∼1.5 μJ for the IR beam. The beam diameters at the foci were approximately 50 μm. The SFG spectra were normalized using the SFG response from a layer of gold on fused silica to account for the distribution of IR energy. The spectra were then calibrated to the methyl C–H stretches of a polystyrene spectroscopic standard and then averaged over two to four laser spots taken on the same sample.52 The SFG spectra were collected from particle filter samples that were pressed with a fused silica window under conditions approaching total internal reflection. In this work, we utilized the ssp polarization combination. In this polarization combination, the upconverter and infrared light fields are plane-polarized parallel (s) and perpendicular (p) to the surface, respectively, and the SFG light field is plane-polarized parallel (s) to the surface, to probe vibrations perpendicular to the surface.
:50 beam splitter, and half was used to pump an optical parametric amplifier (Spectra Physics OPA 800C) which generated broadband IR pulses centered at ∼3.4 μm. SFG spectra were collected using a hybrid scanning/broadband method pioneered by Esenturk and Walker.51 Using this method, the IR light field was incident on the sample at several center wavelengths so that the full frequency range of interest was subjected to approximately the same incident IR power. This method allowed us to cover the CH stretching frequency region between 2700 and 3200 cm−1. To prevent optical damage to the samples, the incident pulse energies were limited to 1 μJ for the 800 nm beam and ∼1.5 μJ for the IR beam. The beam diameters at the foci were approximately 50 μm. The SFG spectra were normalized using the SFG response from a layer of gold on fused silica to account for the distribution of IR energy. The spectra were then calibrated to the methyl C–H stretches of a polystyrene spectroscopic standard and then averaged over two to four laser spots taken on the same sample.52 The SFG spectra were collected from particle filter samples that were pressed with a fused silica window under conditions approaching total internal reflection. In this work, we utilized the ssp polarization combination. In this polarization combination, the upconverter and infrared light fields are plane-polarized parallel (s) and perpendicular (p) to the surface, respectively, and the SFG light field is plane-polarized parallel (s) to the surface, to probe vibrations perpendicular to the surface.
3. Results and discussion
3.1 Morphology and elemental analysis of sea spray aerosol particles
Fig. 1 shows a typical optical microscope image of SSA particles in the supermicrometer size range, collected on a substrate under ambient conditions. Several particles contain a cube-shaped NaCl crystal that can be seen when the tops of the particles are in focus (Fig. 1a). The particles were likely aqueous when they were collected, because the 60% relative humidity (RH) in the wave flume exceeded the efflorescence RH of NaCl at room temperature. The crystal is present because the location of the laboratory microscope and storage had an RH of <35%, lower than the efflorescence RH of NaCl (40%).53 When the microscope focus was set at the substrate instead of the top of the particle, coatings surrounding these NaCl crystallites were easily seen (Fig. 1b). Prior work shows this coating consists of organic species,4,5,16,22 sulfates (i.e. CaSO4 and Na2SO4),43 carbonates (i.e. CaCO3),54 and other hygroscopic salts, e.g. MgCl2, CaCl2, or KMgCl3·6H2O.43,55 | ||
| Fig. 1 Optical microscope images of particles in the supermicrometer range, showing the NaCl crystallite and the coatings that surround these sea spray particles; (a) the microscope is focused on the top of the particles; (b) the microscope is focused on the bottom of the particles where coatings can be more easily seen. | ||
In order to gain additional insights into the morphology and elemental composition of the SSA particles studied here, particles were characterized with SEM-EDX (Fig. 2) under vacuum conditions. As observed with the optical image in Fig. 1, the particles are nearly cubic in morphology, with a coating that has a diffuse appearance in some locations and rod-like character in others. EDX spectra of different regions of the particle within the cube show NaCl as the major component. When the electron beam was focused on the peripheral portion of the particle, the relative intensities of the following elements, Mg, Ca, C, O, and S, increased significantly compared to the levels of Na and Cl in the cubic portion. These findings are consistent with the previously identified major inorganic elemental components of SSA particles and seawater.2 For example, rod-shaped crystals attached to sea salt particles have been reported in other studies and are assigned to crystalline CaSO4.56,57 Moreover, the detected carbon must be attributed to organic species, because Raman microspectroscopy reveals that carbonates are rarely present in the particles (vide infra). The Raman spectrum associated with the same particle shown in Fig. 2 is included in the ESI† as Fig. S1, which also shows no carbonate present, as does the more extensive Raman microspectroscopy data presented in the next section.
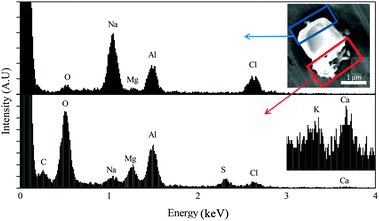 | ||
| Fig. 2 SEM image (upper right) of a sea spray aerosol particle composed mainly of salts. EDX spectra for two areas of the particle (indicated by rectangles) are shown in the upper and lower panels. The accelerating voltage was 5 kV. The inset in the bottom panel shows a magnified spectrum obtained with 7.5 kV accelerating voltage to amplify signals from potassium and calcium. Al is due to the substrate and not present in the sample. | ||
3.2 Raman microspectroscopy of individual substrate-deposited sea spray particles
Fig. 3 shows a typical Raman spectrum of an individual SSA particle (2 μm diameter), the expanded C–H stretching region, and a Raman spectrum of the fused quartz substrate. The substrate contributes only minimally to the spectrum of the SSA, and its contribution is simple. Based on Raman spectra of reference compounds (Fig. S2, ESI†), as well as reports in the literature,58–60 the peak at 1010 cm−1 is assigned as the symmetric stretch of sulfate, ν1(SO42−), likely gypsum (CaSO4·2H2O), though the specific form of calcium sulfate will depend on the conditions of the efflorescence process and can exist as bassanite (CaSO4·0.5H2O).55,61,62 The peak observed at 1050 cm−1 is due to the ν1(HSO4−) mode, specifically the symmetric stretch of the SO3 subgroup.60,63–65 A weaker sulfate signal at 1140 cm−1 is also observed for samples with a strong ν1(SO42−) mode, resulting from the ν3(SO42−) mode, which also matches the frequency for the ν3(SO42−) mode of gypsum.43,61,62 While other studies have suggested that CaCO3 should be prevalent in the SSA, the symmetric stretch of ν1(CO32−) at 1090 cm−1 was only observed in a low percentage of SSA particles.66,67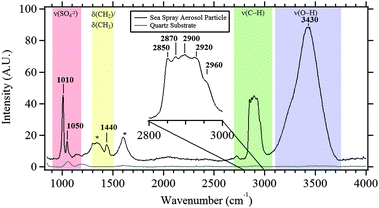 | ||
| Fig. 3 Typical Raman spectra of the quartz substrate and of a 2 μm sea spray aerosol particle (with inset of C–H stretching region). Bands associated with various functional groups are labeled (see text for details). *These bands are due to graphitic carbon that forms due to laser burning of the sample. | ||
Another pronounced feature in the Raman spectrum shown in Fig. 3 is a broad peak(s) between 3200 and 3700 cm−1, which may be primarily assigned to O–H stretches of water, with minor contributions from organic hydroxyl groups, including alcohol groups. Fig. 4 shows six different particles in the C–H stretching and O–H stretching regions. There is variability across particles. For example, in Fig. 4b, e and f, the broad band in the O–H stretching region has more structure and narrower, more well-defined peaks due to crystalline water. This is most likely due to water in divalent salts including MgCl2/CaCl2.53,68 For particles with high sulfate signal, structure is observed in the O–H stretching region with peaks in different particles attributable, according to literature values, to CaSO4·2H2O (3405 and 3495 cm−1), CaSO4·0.5H2O (3510 cm−1), MgSO4 (3440 cm−1), along with MgCl2 and CaCl2 in H2O (3233 cm−1),69 and KMgCl3·6H2O (3259 and 3430 cm−1).43,55,70
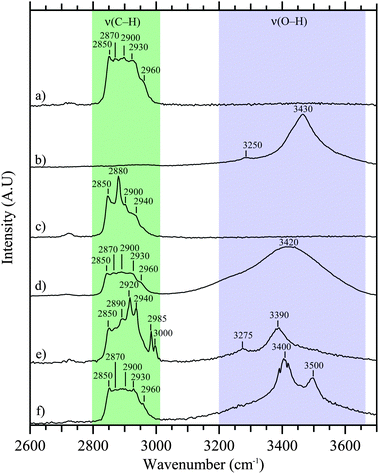 | ||
| Fig. 4 Raman spectra of six different sea spray aerosol particles in the C–H and O–H stretching regions. Particles (a–c) were collected before addition of biological and organic material, while particles (d–f) were collected after addition. | ||
Raman peaks in the aliphatic C–H stretching region are also prominent. The breadth and featureless nature of some of the Raman spectra suggest there are several organic species contributing to this spectral region. Peaks at 2850 cm−1 and 2930 cm−1 can be assigned to the symmetric and asymmetric stretches of C–H in methylene groups, respectively,71 whereas peaks at 2870 cm−1 and 2960 cm−1 can be assigned to the symmetric and asymmetric stretches of C–H in methyl groups, respectively.71 These spectral signatures are consistent with the Raman spectra of various functionalized organic reference compounds for organic surfactants and known oceanic species, including sodium dodecyl sulfate (SDS), CH3(CH2)11OSO3Na; palmitic acid, CH3(CH2)14COOH; glycine, NH2CH2COOH; and lipopolysaccharides (LPS) from Escherichia Coli, presented and discussed in the ESI† (Fig. S3).71 Additionally, the peak at 1440 cm−1 is associated with the bending mode of CH2/CH3 groups (labeled as δ(CH2)/δ(CH3)). Broad peaks at 1350 and 1590 cm−1 are consistent with damage from the laser beam and the formation of graphitic carbon.72
The variability of different spectral features seen in the C–H stretching region is shown in Fig. 4. The frequencies of the C–H stretching modes are below 3000 cm−1, consistent with their assignment to saturated aliphatic hydrocarbons. In general, the spectral variability observed in the Raman spectra of individual SSA particles is attributed to the presence of several organic components in and on the particles. Linear combinations of some of the organic reference compounds, all of which are highly aliphatic in nature, have similar characteristics of the peak frequencies and intensities observed for SSA particles, suggesting a mixed organic layer of several compounds. Fig. 5 shows simulated spectra of linear combinations for selected organic standards that have been associated with organics in seawater73–75 and contain qualitative similarities to the C–H stretching region shown in Fig. 3 (and Fig. 4a, d and f), which shows the most commonly observed C–H region for SSA particles analyzed here with Raman microspectroscopy. The residual was minimized between the spectrum in Fig. 3 and different linear combinations: LPS and SDS (Fig. 5a); SDS and glycine (Fig. 5b); LPS and glycine (Fig. 5c); and LPS, SDS, and glycine (Fig. 5d). Residual values were higher with the fourth organic standard (palmitic acid) and are not shown. The lowest residual was for a combination of 20% LPS, 50% SDS, and 30% glycine. Thus, these measurements suggest the most abundant organic species contain aliphatic moieties and are multicomponent in nature comprised of previously detected oceanic organic species.71
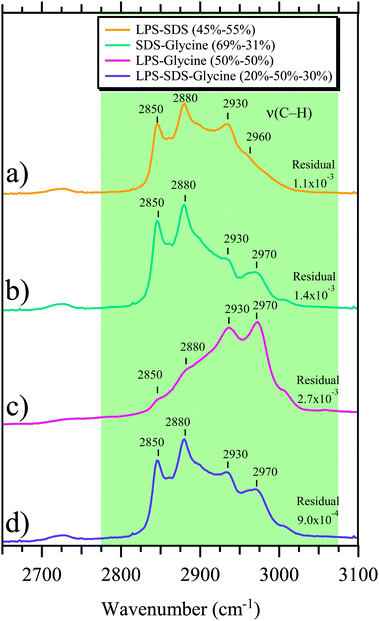 | ||
| Fig. 5 Simulated Raman spectra are shown in the C–H stretching region. These simulated spectra represent different combinations of spectral weighting of three different select organic standards (sodium dodecyl sulfate (SDS) – CH3(CH2)11OSO3Na, lipopolysaccharides from Escherichia Coli (LPS) and Glycine (–NH2CH2COOH). The spectral weighting of each component is given in the inset for the four different simulated Raman spectra. The four combinations shown are a result of minimizing the residual when compared to the SSA Raman spectrum shown in Fig. 3 for: (a) SDS and LPS, (b) SDS and Glycine, (c) LPS and Glycine, and (d) LPS, SDS, and Glycine. | ||
For samples measured before the addition of bacteria and phytoplankton to the sea water in the wave flume, a small fraction of particles exhibit higher intensity in the peaks at 2850 and 2880 cm−1 (Fig. 4c). After bacteria and phytoplankton were added, a small fraction of particles exhibit increased C–H stretching modes in the range from 2920 to 2940 cm−1, with new peaks in the range of 2985 to 3000 cm−1, as in Fig. 4e. These new peaks may indicate the presence of species due to the bacteria and/or phytoplankton added to the sea water.45
The particles observed here can be organized into two groups, and following Prather et al., we categorize them as Type 1 (designated SS for sea salt) and Type 2 (designated SS-OC for sea salt with organic compounds).45 It is clear that there is a transition from Type 1 to Type 2 particles as a function of decreasing particle size. To better view the transition from particles dominated by inorganic ions (in this case as measured by the presence of sulfate ion) with little organic matter to particles that contained significant amounts of organic matter, the integrated area of the Raman peaks were determined for ∼20 particles on each MOUDI stage probed by Raman microspectroscopy (97 total). Raman spectra of individual particles across the size range measured are shown in Fig. S4 of the ESI.† The average areas were then determined for the ν(C–H) (2800 to 3000 cm−1) mode and the ν1(SO42−) (980 to 1030 cm−1) mode, normalized to the highest value, and plotted as a function of particle size in Fig. 6a. The relative intensity of the signals for Raman peaks associated with organic matter is clearly seen to increase with decreasing size, despite lower overall spectral intensity with smaller particles. A corresponding decrease in sulfate intensity is observed with decreasing size.
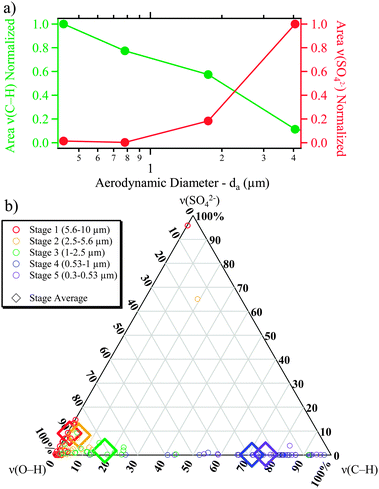 | ||
| Fig. 6 (a) Normalized integrated area of Raman bands for ν(SO42−) and ν(C–H) modes, as a function of particle size. (b) Ternary plot showing the relative areas of the ν(SO42−), ν(C–H), ν(O–H) modes. For the ternary plot, color represents the particle stage, the diamonds represent the average values for a stage, while the smaller circles represent individual particles. | ||
To further relate the three vibrational regions for particles across the size range analyzed, a ternary plot of integrated peak areas for individual particles, along with averages by stage, is shown in Fig. 6b for the ν(C–H), the ν(SO42−), and the ν(O–H) modes with color indicating the stage on which the particles were collected. The ν(O–H) modes include both crystalline and aqueous water associated with the particles as well as any other species with an O–H group. A clear shift is observed from Raman spectra dominated by the ν1(SO42−) and the ν(O–H) modes at larger particle sizes to spectra dominated by ν(C–H) modes at smaller particle sizes. The high organic matter content demonstrated in Fig. 6 for the submicrometer-sized particles has important implications for the water uptake properties and heterogeneous reactivity of these particles, which is further explored with SFG in the following section.
3.3 Organic species detected at the surfaces and interfaces of SSA particles
The interface between the particle surface and gas phase is where many of the chemical and physical interactions occur that ultimately determine the climate properties of SSA particles. Given the substantial role that ocean biological activity can play in affecting the bulk chemical composition of SSA particles, SSA particles were investigated using vibrational SFG in the C–H stretching region. SFG signals can only be produced by C–H oscillators located in non-centrosymmetric environments, such as at the external surface of a homogeneous aerosol particle. An oscillator located at the internal interface separating two immiscible centrosymmetric phases within an aerosol particle can also contribute to the SFG response. This requirement distinguishes the method from other vibrational spectroscopic methods used for aerosol particle analysis, in which the oscillators of all species present contribute. Of those many species, only the oscillators located at the surface of a given particle and interfaces within it, that are also part of a non-zero average net orientation distribution on that particle surface can produce the ssp-polarized SFG responses discussed here. The collective SFG response from the multiple particles present in the laser spot scales quadratically with the number of oscillators on each particle surface but linearly with the number of particles in the laser spot.76All particle-containing samples that were investigated by SFG show signal intensity at approximately 2850 and 2920 cm−1, consistent with the presence of methylene symmetric and asymmetric stretching modes, respectively, and 2880 and 2950 cm−1, consistent with the presence of methyl symmetric and asymmetric stretching modes, respectively.77–83 The mode near 2950 cm−1 may also have contributions from methyl Fermi resonances. While significant signal intensity exists around 2900 cm−1, spectral features are not particularly well resolved for these samples in that frequency region. The samples also show weak and broad SFG responses in the 3100 cm−1 to 3200 cm−1 region, typically associated with the presence of O–H stretches. Control studies (not shown) indicate that signal contributions from adventitious carbon material that might be present on aerosol particle surfaces are minor.84
The SFG responses of the particles in the supermicrometer size range are dominated by vibrational resonances at 2880 cm−1 and 2950 cm−1 (Fig. 7a). In addition to these two spectral features, the SFG responses of the particles also exhibit vibrational resonances at 2850 cm−1 and 2920 cm−1 (Fig. 7a), which begin to be resolved to some degree with decreasing size as shown in spectra of particles having aerodynamic diameters of 150 nm. For seawater containing added bacteria and phytoplankton (Fig. 7b), the ratio of the SFG intensities of the vibrational modes at 2880 and 2950 cm−1 decreases from 1:2 to 1:1 for particles having diameters greater than 1 μm. By contrast, the relative intensities of these bands do not change significantly for the 150 nm particles. As noted above, spectral features at 2880 and 2950 cm−1 are commonly attributed to methyl symmetric and asymmetric stretching modes, respectively. The relative ratio of SFG response at these two frequencies is commonly understood to be related to the molecular orientation of the methyl groups sampled.84 A change in the ratio of the SFG responses at these two frequencies has been interpreted, for systems ranging from ordered alkyl chains to polymeric materials at interfaces, to indicate molecular reorientation of the methyl groups towards or away from the surface normal.84 A similar explanation could explain the series of SFG spectra presented here, although a definitive conclusion is not possible given that spectral congestion also affects the appearance of the bands. Without knowing the chemical composition of the particle surfaces sampled here by SFG spectroscopy, it is difficult to go beyond a descriptive level in the discussion rationalizing the changes in the spectral line shapes.
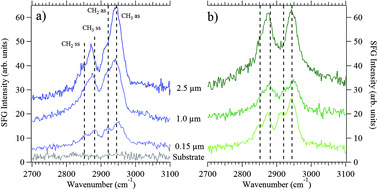 | ||
| Fig. 7 Vibrational SFG spectra (ssp polarization) of sea spray aerosol particles (a) before addition of bacteria and phytoplankton; (b) after addition of bacteria and phytoplankton. The diameters of the particles are indicated in each panel, and spectra are offset for clarity. Vertical dashed lines indicate symmetric and asymmetric stretches, labeled ss and as, respectively, of methyl and methylene groups typically observed for long-chain hydrocarbon systems. Please see text for details. The lowest spectrum in (a) is that of a bare Teflon filter. All of the spectra were recorded under identical spectral acquisition conditions. | ||
While it is not possible at this time to determine (i) the absolute number of C–H oscillators that give rise to the vibrational SFG signals or (ii) the exact chemical composition of organic species producing SFG intensity, the spectra shown in Fig. 7a and b clearly show that C–H oscillators are present at the surfaces of 0.15–2.5 μm aerosol particles collected at the wave flume (and the internal interfaces in the case of mixed-phase particles), even before bacteria and phytoplankton were added to it. We also find that the SFG intensities obtained from the particles collected before and days after addition of bacteria and phytoplankton to the wave flume are comparable within a factor of two, indicating that C–H oscillators are present at the particle surfaces under both conditions.
For each of the samples studied here, the vibrational SFG responses do not indicate the presence of well-ordered alkyl chains. Those well-ordered systems, such as octadecyltrichlorosilane on silica,85 exhibit just two vibrational SFG peaks at 2880 cm−1 and 2950 cm−1, attributable to the methyl symmetric and asymmetric stretches, respectively. Due to local inversion symmetry, those well-ordered systems show only negligible contributions from the symmetric or asymmetric stretching modes of the trans-configured methylene groups and their Fermi resonances, which typically occur at 2850 and 2920 or 2900 cm−1, respectively.77,80 Thus, the SFG spectra reported here are consistent with the hypothesis that the surfaces and internal interfaces of the submicrometer-sized SSA particles contain a significant number of cis-configured methylene oscillators that are somewhat resolved in the SFG spectra, which is in contrast to the SFG spectra obtained from the supermicrometer-sized particles. Ongoing work focuses on developing a molecular level understanding of the SFG responses obtained from the SSA particles through a combination of molecular dynamics simulations, spectroscopy, and climate property measurements.
4. Conclusion and implications
SSA particles formed through wave breaking across the earth's oceans represent the largest flux of aerosol particles generated globally and one of the highest mass concentrations in the atmosphere.3 As SSA particles are complex mixtures consisting of inorganic and organic components that vary as a function of size and seawater composition, simple sizing and elemental analysis is not sufficient to describe SSA particle properties and how they impact climate. Rather, integration of detailed chemical characterization with standard aerosol characterization methods is needed to describe the properties of SSA particles. This study presents the integration of electron microscopy with individual particle (Raman microspectroscopy) and surface-sensitive (SFG) vibrational spectroscopy techniques. These complementary methods provide details on the chemical nature of the organic matter present, in particular for the C–H stretching region. Raman and SFG spectroscopy show C–H stretching modes consistent with aliphatic hydrocarbons. Comparison of SSA across the size range shows that organic matter in individual submicrometer particles exhibited a greater enrichment compared to supermicrometer particles. On the surface of the particles, and at the interfaces within them, organic matter is always present in both submicrometer and supermicrometer particles. Comparison of SSA from seawater prior to and after additions of bacteria and phytoplankton shows changes to the organic matter in the particle phase. SFG spectra of SSA particles after addition are consistent with the notion that the methyl groups reorient. SFG signal for all particles both prior to and after the addition of bacteria and phytoplankton did not indicate a well ordered alkyl chain layer, which has important implications for how these particles take up water and undergo heterogeneous reactivity with trace atmospheric gases, such as N2O5 (g) and HNO3 (g). Taken together, these results show the importance of increasing our understanding of the organic matter portion of SSA particles in the bulk and at particle surfaces under a variety of seawater conditions. This is particularly important as the changes observed in the organic concentration and chemical properties will likely directly impact water uptake, freezing, and light scattering by SSA particles. Further investigations using Raman and SFG are needed to deconvolute the surface coverage of organic matter and the degree to which it is ordered, to assess the contribution from internal interfaces versus the external surface of a mixed-phase particle, as well as to directly study different mixtures of biological species to correlate with specific spectroscopic signatures. These results set the stage for future efforts to describe SSA particles in a manner that can be linked with reactivity and climate relevant properties.Acknowledgements
This study is based upon work supported by the National Science Foundation through a Phase I Center for Chemical Innovation under Grant No. CHE1038028. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The authors thank all those involved with the CAICE intensive campaign, including: Dr Luis Cuadra-Rodriguez, Dr Timothy Guasco, Douglas Collins, Matt Ruppel, Prof. Timothy Bertram, Michelle Kim, Olivia Ryder, Dr Grant Deane, Dr Dale Stokes, Prof. Farooq Azam, and the SIO Hydraulics Laboratory staff. The authors wish to thank Camille Sultana and Chen Wang for their assistance with the initial Raman analysis. We gratefully acknowledge the loan of a Raman spectrometer to obtain initial data that was provided by Dr Andrew King and Renishaw and the assistance of Mr Ryan Anderson in NANO3, CALIT2, University of California, San Diego in the SEM use. CJE gratefully acknowledges an NSF Graduate Research Fellowship.References
- G. de Leeuw, E. L. Andreas, M. D. Anguelova, C. W. Fairall, E. R. Lewis, C. O'Dowd, M. Schulz and S. E. Schwartz, Rev. Geophys, 2011, 49 DOI:10.1029/2010rg000349.
- R. Lewis and E. Schwartz, Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models – A Critical Review, AGU, Washington, DC, 2004 Search PubMed.
- M. O. Andreae and D. Rosenfeld, Earth-Sci. Rev., 2008, 89, 13–41 CrossRef.
- C. D. O'Dowd, M. C. Facchini, F. Cavalli, D. Ceburnis, M. Mircea, S. Decesari, S. Fuzzi, Y. J. Yoon and J. P. Putaud, Nature, 2004, 431, 676–680 CrossRef CAS.
- D. M. Murphy, J. R. Anderson, P. K. Quinn, L. M. McInnes, F. J. Brechtel, S. M. Kreidenweis, A. M. Middlebrook, M. Posfai, D. S. Thomson and P. R. Buseck, Nature, 1998, 392, 62–65 CrossRef CAS.
- D. A. Knopf, P. A. Alpert, B. Wang and J. Y. Aller, Nat. Geosci., 2011, 4, 88–90 CrossRef CAS.
- C. D. O'Dowd and G. De Leeuw, Philos. Trans. R. Soc. London, Ser. A, 2007, 365, 1753–1774 CrossRef CAS.
- L. I. Aluwihare and D. J. Repeta, Mar. Ecol.: Prog. Ser., 1999, 186, 105–117 CrossRef CAS.
- P. Verdugo, A. L. Alldredge, F. Azam, D. L. Kirchman, U. Passow and P. H. Santschi, Mar. Chem., 2004, 92, 67–85 CrossRef CAS.
- M. V. Orellana, P. A. Matrai, C. Leck, C. D. Rauschenberg, A. M. Lee and E. Coz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13612–13617 CrossRef CAS.
- E. C. Griffith, A. F. Tuck and V. Vaida, Acc. Chem. Res., 2012, 45, 2106–2113 CrossRef CAS.
- D. J. Donaldson and V. Vaida, Chem. Rev., 2006, 106, 1445–1461 CrossRef CAS.
- D. C. Blanchard, Science, 1964, 146, 396–397 CAS.
- G. B. Ellison, A. F. Tuck and V. Vaida, J. Geophys. Res., [Atmos.], 1999, 104, 11633–11641 CrossRef CAS.
- F. Cavalli, M. C. Facchini, S. Decesari, M. Mircea, L. Emblico, S. Fuzzi, D. Ceburnis, Y. J. Yoon, C. D. O'Dowd, J. P. Putaud and A. Dell'Acqua, J. Geophys. Res., [Atmos.], 2004, 109 DOI:10.1029/2004JD005137.
- M. C. Facchini, M. Rinaldi, S. Decesari, C. Carbone, E. Finessi, M. Mircea, S. Fuzzi, D. Ceburnis, R. Flanagan, E. D. Nilsson, G. de Leeuw, M. Martino, J. Woeltjen and C. D. O'Dowd, Geophys. Res. Lett., 2008, 35 DOI:10.1029/2008GL034210.
- D. Ceburnis, A. Garbaras, S. Szidat, M. Rinaldi, S. Fahrni, N. Perron, L. Wacker, S. Leinert, V. Remeikis, M. C. Facchini, A. S. H. Prevot, S. G. Jennings, M. Ramonet and C. D. O'Dowd, Atmos. Chem. Phys., 2011, 11, 8593–8606 CrossRef CAS.
- E. Fuentes, H. Coe, D. Green and G. McFiggans, Atmos. Chem. Phys., 2011, 11, 2585–2602 CrossRef CAS.
- E. Gard, J. E. Mayer, B. D. Morrical, T. Dienes, D. P. Fergenson and K. A. Prather, Anal. Chem., 1997, 69, 4083–4091 CrossRef CAS.
- A. P. Ault, T. M. Peters, E. J. Sawvel, G. S. Casuccio, R. D. Willis, G. A. Norris and V. H. Grassian, Environ. Sci. Technol., 2012, 46, 4331–4339 CrossRef CAS.
- C. Leck and E. K. Bigg, Geophys. Res. Lett., 2005, 32 DOI:10.1029/2005gl023651.
- E. K. Bigg and C. Leck, J. Geophys. Res., [Atmos.], 2008, 113 DOI:10.1029/2007jd009078.
- C. J. Gaston, H. Furutani, S. A. Guazzotti, K. R. Coffee, T. S. Bates, P. K. Quinn, L. I. Aluwihare, B. G. Mitchell and K. A. Prather, J. Geophys. Res., [Atmos.], 2011, 116 DOI:10.1029/2010jd015289.
- H. Mouri, K. Okada and K. Shigehara, Tellus, Ser. B, 1993, 45, 80–85 CrossRef.
- H. Geng, F. Q. Cheng and C. U. Ro, J. Air Waste Manage. Assoc., 2011, 61, 1183–1191 CAS.
- H. Geng, S. Kang, H. J. Jung, M. Choel, H. Kim and C. U. Ro, J. Geophys. Res., [Atmos.], 2010, 115 DOI:10.1029/2009jd013486.
- L. N. Hawkins and L. M. Russell, Adv. Met., 2010 DOI:10.1155/2010/612132.
- Y. C. Song, J. Ryu, M. A. Malek, H. J. Jung and C. U. Ro, Anal. Chem., 2010, 82, 7987–7998 CrossRef CAS.
- E. E. Gard, M. J. Kleeman, D. S. Gross, L. S. Hughes, J. O. Allen, B. D. Morrical, D. P. Fergenson, T. Dienes, M. E. Galli, R. J. Johnson, G. R. Cass and K. A. Prather, Science, 1998, 279, 1184–1187 CrossRef CAS.
- A. Worobiec, S. Potgieter-Vermaak, A. Brooker, L. Darchuk, E. Stefaniak and R. Van Grieken, Microchem. J., 2010, 94, 65–72 CrossRef CAS.
- J. Y. Yu, Y. Zhang, G. Zeng, C. M. Zheng, Y. Liu and Y. H. Zhang, J. Phys. Chem. B, 2012, 116, 1642–1650 CrossRef CAS.
- V. G. Ciobanu, C. Marcolli, U. K. Krieger, U. Weers and T. Peter, J. Phys. Chem. A, 2009, 113, 10966–10978 CrossRef CAS.
- J. Buajarern, L. Mitchem and J. P. Reid, J. Phys. Chem. A, 2007, 111, 9054–9061 CrossRef CAS.
- A. K. Y. Lee and C. K. Chan, Atmos. Environ., 2007, 41, 4611–4621 CrossRef CAS.
- M. E. Wise, K. J. Baustian and M. A. Tolbert, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6693–6698 CrossRef CAS.
- Y. J. Liu, T. Zhu, D. F. Zhao and Z. F. Zhang, Atmos. Chem. Phys., 2008, 8, 7205–7215 CrossRef CAS.
- N. Kwamena and R. Jonathan, Fundamentals and Applications in Aerosol Spectroscopy, CRC Press, 2010, pp. 127–153 Search PubMed.
- K. H. Fung, Chemical characterization of aerosol particles by laser Raman spectroscopy, Brookhaven National Laboratory, Upton, NY (US), 1999 Search PubMed.
- K. J. Baustian, D. J. Cziczo, M. E. Wise, K. A. Pratt, G. Kulkarni, A. G. Hallar and M. A. Tolbert, J. Geophys. Res., [Atmos.], 2012, 117 DOI:10.1029/2011jd016784.
- Y. Batonneau, S. Sobanska, J. Laureyns and C. Bremard, Environ. Sci. Technol., 2006, 40, 1300–1306 CrossRef CAS.
- K. Deboudt, P. Flament, M. Choel, A. Gloter, S. Sobanska and C. Colliex, J. Geophys. Res., [Atmos.], 2010, 115 DOI:10.1029/2010jd013921.
- S. Sobanska, H. Hwang, M. Choel, H. J. Jung, H. J. Eom, H. Kim, J. Barbillat and C. U. Ro, Anal. Chem., 2012, 84, 3145–3154 CrossRef CAS.
- H. S. Xiao, J. L. Dong, L. Y. Wang, L. J. Zhao, F. Wang and Y. H. Zhang, Environ. Sci. Technol., 2008, 42, 8698–8702 CrossRef CAS.
- C. J. Ebben, I. S. Martinez, M. Shrestha, A. M. Buchbinder, A. L. Corrigan, A. Guenther, T. Karl, T. Petaja, W. W. Song, S. R. Zorn, P. Artaxo, M. Kulmala, S. T. Martin, L. M. Russell, J. Williams and F. M. Geiger, Atmos. Chem. Phys., 2011, 11, 10317–10329 CrossRef CAS.
- K. A. Prather, T. H. Bertram, V. H. Grassian, F. M. Geiger, G. B. Deane, M. D. Stokes, P. J. DeMott, L. I. Aluwihare, F. Azam, J. H. Seinfeld, R. C. Moffet, M. J. Molina, C. D. Cappa, F. M. Geiger, G. C. Roberts, L. M. Russell, A. P. Ault, J. Baltrusaitis, L. A. Caudra-Rodriguez, D. B. Collins, C. E. Corrigan, S. D. Forestieri, C. J. Ebben, T. L. Gausco, S. P. Hersey, M. J. Kim, R. L. Modini, W. Mu, B. E. Pedler, M. J. Ruppel, O. S. Ryder, N. Schoepp, R. C. Sullivan and D. Zhao, 2013, submitted.
- http://caice.ucsd.edu/ .
- G. B. Deane and M. D. Stokes, Nature, 2002, 418, 839–844 CrossRef CAS.
- C. J. Ebben, M. Shrestha, I. S. Martinez, A. L. Corrigan, A. A. Frossard, W. W. Song, D. R. Worton, T. Petäjä, J. Williams, L. M. Russell, M. Kulmala, A. H. Goldstein, P. Artaxo, S. T. Martin, R. J. Thomson and F. M. Geiger, J. Phys. Chem. A, 2012, 116, 8271–8290 CrossRef CAS.
- A. B. Voges, H. A. Al-Abadleh and F. M. Geiger, in Environmental Catalysis, ed. V. H. Grassian, CRC Press, Boca Raton, 2005 Search PubMed.
- G. Y. Stokes, A. M. Buchbinder, J. M. Gibbs-Davis, K. A. Scheidt and F. M. Geiger, Vib. Spectrosc., 2009, 50, 86–98 CrossRef CAS.
- O. Esenturk and R. A. Walker, J. Phys. Chem. B, 2004, 108, 10631–10635 CrossRef CAS.
- A. B. Voges, H. A. Al-Abadleh, M. J. Musorrariti, P. A. Bertin, S. T. Nguyen and F. M. Geiger, J. Phys. Chem. B, 2004, 108, 18675–18682 CrossRef CAS.
- D. J. Cziczo and J. P. D. Abbatt, J. Phys. Chem. A, 2000, 104, 2038–2047 CrossRef CAS.
- W. C. Keene, H. Maring, J. R. Maben, D. J. Kieber, A. A. P. Pszenny, E. E. Dahl, M. A. Izaguirre, A. J. Davis, M. S. Long, X. Zhou, L. Smoydzin and R. Sander, J. Geophys. Res., [Atmos.], 2007, 112 DOI:10.1029/2007jd008464.
- H. J. Tong, Z. G. Qian, J. P. Reid and Y. H. Zhang, Acta Phys.-Chim. Sin., 2011, 27, 2521–2527 CAS.
- M. E. Wise, T. A. Semeniuk, R. Bruintjes, S. T. Martin, L. M. Russell and P. R. Buseck, J. Geophys. Res., [Atmos.], 2007, 112 DOI:10.1029/2006JD007678.
- M. M. Rashad, M. H. H. Mahmoud, I. A. Ibrahim and E. A. Abdel-Aal, J. Cryst. Growth, 2004, 267, 372–379 CrossRef CAS.
- L. P. Sarma, P. S. R. Prasad and N. Ravikumar, J. Raman Spectrosc., 1998, 29, 851–856 CrossRef CAS.
- Y. H. Zhang and C. K. Chan, J. Phys. Chem. A, 2000, 104, 9191–9196 CrossRef CAS.
- F. P. Daly, D. R. Kester and C. W. Brown, J. Phys. Chem., 1972, 76, 3664–3668 CrossRef CAS.
- P. V. Jentzsch, R. M. Bolanz, V. Ciobota, B. Kampe, P. Rosch, J. Majzlan and J. Popp, Vib. Spectrosc., 2012, 61, 206–213 CrossRef.
- B. J. Berenblut, P. Dawson and G. R. Wilkinson, Spectrochim. Acta, Part A, 1973, 29, 29–36 CrossRef CAS.
- V. Vchirawongkwin, C. Kritayakornupong and B. M. Rode, J. Phys. Chem. B, 2010, 114, 11561–11569 CrossRef CAS.
- K. H. Fung and I. N. Tang, Appl. Spectrosc., 1991, 45, 734–737 CrossRef CAS.
- R. J. Gillespie and E. A. Robinson, Can. J. Chem., 1962, 40, 644–657 CrossRef CAS.
- F. A. Andersen and L. Brecevic, Acta Chem. Scand., 1991, 45, 1018–1024 CrossRef CAS.
- S. Gunasekaran, G. Anbalagan and S. Pandi, J. Raman Spectrosc., 2006, 37, 892–899 CrossRef CAS.
- D. J. Cziczo, J. B. Nowak, J. H. Hu and J. P. D. Abbatt, J. Geophys. Res., [Atmos.], 1997, 102, 18843–18850 CrossRef CAS.
- R. H. Li, Z. P. Jiang, S. Q. Shi and H. W. Yang, J. Mol. Struct., 2003, 645, 69–75 CrossRef CAS.
- F. Wang, Y. H. Zhang, S. H. Li, L. Y. Wang and L. J. Zhao, Anal. Chem., 2005, 77, 7148–7155 CrossRef CAS.
- P. Larkin, Infrared and Raman Spectroscopy; Principles and Spectral Interpretation, Elsevier, 2011 Search PubMed.
- S. Mertes, B. Dippel and A. Schwarzenbock, J. Aerosol Sci., 2004, 35, 347–361 CrossRef CAS.
- J. D. Pakulski and R. Benner, Limnol. Oceanogr., 1994, 39, 930–940 CrossRef CAS.
- R. N. Roslan, N. M. Hanif, M. R. Othman, W. Azmi, X. X. Yan, M. M. Ali, C. A. R. Mohamed and M. T. Latif, Mar. Pollut. Bull., 2010, 60, 1584–1590 CrossRef CAS.
- P. Schmitt-Kopplin, G. Liger-Belair, B. P. Koch, R. Flerus, G. Kattner, M. Harir, B. Kanawati, M. Lucio, D. Tziotis, N. Hertkorn and I. Gebefuegi, Biogeosciences, 2012, 9, 1571–1582 CrossRef CAS.
- E. C. Y. Yan, Y. Liu and K. B. Eisenthal, J. Phys. Chem. B, 2001, 105, 8531–8537 CrossRef CAS.
- Y. R. Shen, Nature, 1989, 337, 519–525 CrossRef CAS.
- Y. Liu, L. K. Wolf and M. C. Messmer, Langmuir, 2001, 17, 4329–4335 CrossRef CAS.
- C. Y. Chen, C. L. Loch, J. Wang and Z. Chen, J. Phys. Chem. B, 2003, 107, 10440–10445 CrossRef CAS.
- S. Nihonyanagi, D. Miyamoto, S. Idojiri and K. Uosaki, J. Am. Chem. Soc., 2004, 126, 7034–7040 CrossRef CAS.
- D. L. Elmore, C. L. Leverette, D. B. Chase, A. T. Kalambur, Y. J. Liu and J. F. Rabolt, Langmuir, 2003, 19, 3519–3524 CrossRef CAS.
- R. G. Snyder, H. L. Strauss and C. A. Elliger, J. Phys. Chem., 1982, 86, 5145–5150 CrossRef CAS.
- R. G. Snyder, S. L. Hsu and S. Krimm, Spectrochim. Acta, Part A, 1978, 34, 395–406 CrossRef.
- J. Wang, Z. Paszti, M. A. Even and Z. Chen, J. Am. Chem. Soc., 2002, 124, 7016–7023 CrossRef CAS.
- S. R. Walter, J. Youn, J. D. Emery, S. Kewalramani, J. W. Hennek, M. J. Bedzyk, A. Facchetti, T. J. Marks and F. M. Geiger, J. Am. Chem. Soc., 2012, 134, 11726–11733 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp43899f |
| ‡ Authors with equal contributions. |
| § Present address: Institute of Energy and Climate: Troposphere (IEK-8), Forschungszentrum Juelich, Juelich, D-52428, Germany. |
| This journal is © the Owner Societies 2013 |