 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of the hydrogen bond in dense nanoparticle–gas suspensions
Maryam
Tahmasebpoor†
a,
Lilian
de Martín†
b,
Mojgan
Talebi
b,
Navid
Mostoufi
a and
J. Ruud
van Ommen
*b
aMultiphase Research Systems Lab., School of Chemical Engineering, College of Engineering, University of Tehran, Tehran, Iran
bDelft University of Technology, Department of Chemical Engineering, Product & Process Engineering, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: J.R.vanOmmen@tudelft.nl; Fax: +31 1527 88267; Tel: +31 1527 82133
First published on 19th February 2013
Abstract
The effect of surface characteristics on the interaction between nanoparticles and their agglomeration in dense gas suspensions is still not fully understood. It is known that when the surface is covered with hydroxyl groups, the interaction between nanoparticles becomes substantially stronger than in the absence of these groups; this strengthening is typically attributed to the formation of capillary bridges between the particles. However, this work shows that part of the increase of the interaction is due to the direct hydrogen bonds formed between the surfaces of the polar particles. Dry nitrogen was used to fluidize polar (hydrophilic) and apolar (hydrophobic) SiO2, TiO2 and Al2O3 particles, with a size ranging from 13 to 21 nm. The dry polar particles showed smaller bed expansion and larger minimum fluidization velocity compared to their apolar counterparts, indicating stronger interparticle forces. The results show the importance of including the formation of hydrogen bonds in the modeling of the interaction between dry and polar nanoparticles.
1 Introduction
Nanoparticles are being used in a number of processes, and their application is increasing because of their unique chemical, electro-magnetic, optical, and other properties resulting from the high surface area to mass ratio. A proper understanding of the interaction between nanoparticles in the gas phase will allow the prediction of the structure and size of their agglomerates, and their interaction with the surrounding media. Such insight is valuable in various fields, such as production processes,1,2 inhalation studies,3 and toxicology analysis.4The attractive forces commonly considered in modeling the interactions between nanoparticles in the gas phase include: (1) van der Waals, (2) electrostatics and (3) capillary forces.2,5,6 Van der Waals forces (FvdW) are the result of interaction between dipoles and are considered dominant in the absence of capillary forces.2 The van der Waals force between two equal smooth spherical particles of size dp is:7
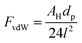 | (1) |
Electrostatics takes place when the charges inside a particle/agglomerate are displaced and the surface charge density changes, a phenomenon known as the patch charge effect.10 The evaluation of the electrostatic interaction between nanoparticles is complex since it depends on the presence of humidity and impurities that may give rise to ionic effects at the interface. Electrostatics is more important in non-conductive materials than in conductive materials. However, it is usually neglected when compared to the van der Waals interaction.5,11
Capillary forces originate from adsorption and condensation of molecules on the particle surface forming liquid bridges between the particles. The surface tension of the liquid and the geometry of the neck formed influence the cohesion force. It is commonly accepted that in the presence of humidity capillary forces have an important contribution to the attraction between nanoparticles with a hydrophilic surface.1,12,13 Hakim et al.14 observed how the fluidization of hydrophilic nanoparticles preheated for two hours at 130 °C gave smaller agglomerates than the fluidization without the preheating step. This reduction in the agglomerate size by heating/drying was explained by a decrease in the particle interaction due to the removal of the liquid bridges between nanoparticles. Recent work by Salameh et al.13 revealed that the contact forces between TiO2 nanoparticles under ambient conditions are dominated by the structure of the water layers adsorbed on the particles.
None of the previously mentioned papers consider the contribution of direct hydrogen bonds between nanoparticles to the total attraction. The strong cohesion between hydrophilic nanoparticles in the presence of ambient water vapor is explained by the formation of capillary bridges between the particles, without considering the formation of direct hydrogen bonds. Moreover, the interaction between hydrophilic particles in dry environments is usually estimated using eqn (1), using the Hamaker coefficient of the materials and ignoring the formation of direct hydrogen bonds.12,15–17 It is surprising that in the field of nanoparticle fluidization the possibility of hydrogen bonds between nanoparticles is ignored whereas in other fields it is not only considered, but manipulated to control the assembly of nanostructures.18,19
In this paper, we show that the formation of direct hydrogen bonds between hydrophilic nanoparticles in dry and dense gas suspensions (‘fluidized beds’) cannot be ignored, as it is commonly done. The hydrogen bonds affect the nature of the nanoparticle agglomerates, and thus influence the dynamics of the suspension. If the formation of direct hydrogen bonds is neglected, the interaction between hydrophilic nanoparticles in dry environments will be strongly underestimated, whereas the capillary forces in the presence of vapour might be overestimated.
2 Experimental methods
The nanopowders were fluidized in a 26 mm i.d. glass column to minimize electrostatics as compared to frequently used perspex columns. High-purity nitrogen was supplied to the bed through a porous plate distributor. To study the effect of masking the hydrogen bridges, isopropanol (ISP) was added to the nitrogen in a number of experiments. This was done by bubbling the nitrogen through a bottle filled with ISP before it entered the bed. To prevent the emission of nanoparticles to the atmosphere, the gas flow leaving the system was cleaned with a two-stage water bubbler and then filtered using a HEPA filter. The pressure drop across the bed was measured using a differential pressure transducer (Validyne Engineering, Model DP15-26) and recorded through the data acquisition system. One of the pressure taps was located in the freeboard and the other 3.5 cm above the distributor.Two variants of silica, alumina and titania, with the untreated surface, containing hydroxyl groups, and with an organic coating were investigated. These variants are described by the manufacturer as hydrophilic and hydrophobic, respectively. Since those terms are used to describe the interaction of materials with water and in this work there was no water present, the terms polar and apolar will be used throughout this work. Polar particles are characterized by a surface containing hydroxyl groups, where the concentration of these groups can vary from one type of material to another.20 Apolar particles are produced by substituting the hydroxyl groups of the polar particles by organic groups in a process called hydrophobization21 (Fig. 1).
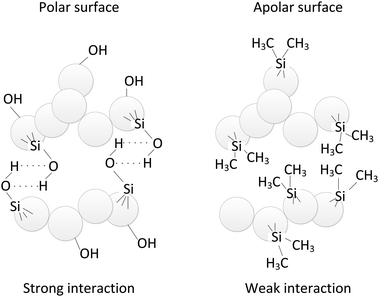 | ||
| Fig. 1 Sketch of the particle–particle interaction for polar and apolar SiO2 nanoparticles. | ||
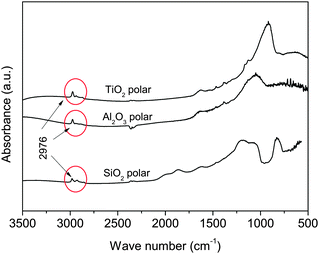
Apolar Aerosil R972 is produced by converting about 30% of the silanol groups present in the polar Aerosil 130 to Si–O–Si–(CH3)2 units.22 In Aeroxide Alu C805, which is the apolar version of Aeroxide Alu C, the hydroxyl groups are substituted by octylsilane-groups H17C8–SiH3.23 Aeroxide T805 is obtained after the adsorption of trimethoxy octyl silane groups CH3(CH2)7Si(OCH3)3 onto the surface of the polar Aeroxide P25.24 Although Aeroxide T805 is hydrophobic, Erdem et al.25 observed that it behaves as if there were still a significant number of reactive hydroxyl groups left on the surface. The same authors quantified the concentration of active hydroxyl groups in Aeroxide P25 and T805 as 3.3 OH per nm2 and 1.8 OH per nm2 respectively.24 FTIR data at 3500–500 cm−1 for the polar and apolar particles as received from the manufacturer are shown in Fig. 2.
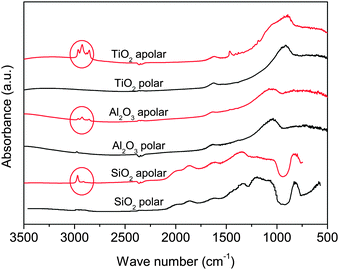 | ||
| Fig. 2 FTIR absorption spectra of the used nanoparticles as received. Circles show the absorption at bands near the stretching vibration of hydrocarbon groups.26 | ||
The main difference between the spectra of polar and apolar particles is in the absorption bands near 2900 cm−1. These peaks are consistent with the stretching vibration of hydrocarbon groups26 and confirm the surface treatment that created the apolar nanoparticles. The properties of the nanopowders used are listed in Table 1. The tapped bulk density was provided by the manufacturer.
| Commercial name | Material | d p [nm] | Surface type | ρ tapped [kg m−3] | ρ p [kg m−3] |
|---|---|---|---|---|---|
| Aerosil 130 | SiO2 | 16 | Polar | 55 | 2200 |
| Aerosil R972 | SiO2 | 16 | Apolar | 85 | 2200 |
| Aeroxide Alu C | Al2O3 | 13 | Polar | 60 | 3600 |
| Aeroxide Alu C805 | Al2O3 | 13 | Apolar | 85 | 3600 |
| Aeroxide P25 | TiO2 | 21 | Polar | 130 | 4000 |
| Aeroxide T805 | TiO2 | 21 | Apolar | 300 | 4000 |
Before the fluidization, the particles were sieved using a 335 μm sieve placed on a shaker. The sieving process removes large agglomerates that may have been generated during packing, storage, and transportation. Sieving, in fact, seems to be an important factor in the ability to fluidize nanoparticles since it removes large particles that encourage channeling.27
The minimum fluidization velocity Umf and the bed expansion were determined to interpret the cohesiveness of the particles. Umf is the lowest velocity at which the particles start to fluidize and can be estimated by measuring the pressure drop through the bed for different gas velocities. Umf is the gas velocity at which an increment in the gas flow does not result in an increment in the pressure drop anymore.28 It is known that the plot of pressure drop vs. gas velocity shows a hysteresis when the gas velocity is increased from a packed bed to a fluidized bed (fluidization) or decreased from a fluidized bed to a packed bed (defluidization).29,30 Since Umf is more reproducible in defluidization,31Umf was calculated by decreasing the superficial gas velocity in small steps of 0.5 mm s−1. After each change in gas velocity, 5 minutes waiting time was taken for the bed to stabilize before the pressure drop was measured.
The dependence of the bed expansion H/H0 on the superficial gas velocity was studied as an indicator of the characteristics of the agglomerates in the bed.1,27H is the height of the bed at a certain superficial gas velocity and H0 is the height of the bed at zero gas velocity. H0 is similar for all the powders and has a value between 5.0 and 5.7 cm.
3 Results and discussion
3.1 Fluidizing with dry nitrogen
Visual observation of the fluidization of the different nanopowders revealed that all particles, except apolar silica, showed what Yao et al.1 denote as agglomerate bubbling fluidization (ABF). ABF behavior is characterized by a nonuniform fluidization with bubbles throughout the bed. The bed expands very little by increasing the gas velocity, and large bubbles rising up very quickly through the bed are observed. The agglomerates are distributed non-uniformly within the bed. Large agglomerates move slowly at the bottom and smaller agglomerates fluidize smoothly in the upper part.1,16In contrast, apolar silica fluidized according to agglomerate particulate fluidization (APF), as previously reported.1 APF behaviour is characterized by homogeneous bubbleless fluidization, where agglomerates are distributed uniformly throughout the bed. With increasing gas velocity, the fluidized bed expands consistently, resulting in a high bed expansion ratio and a fluid-like behavior.1,27 This type of fluidization is associated with low-density nano-powder agglomerates.2
The APF behavior observed for SiO2 agglomerates can be attributed to the low bulk density of this powder. However, the density of the resultant agglomerates is not the only variable affecting the type of fluidization: polar SiO2 has the same particle size and similar bulk density but presents ABF behavior. Since the fluidizing gas in this work was dry nitrogen, the difference in the type of fluidization can only be explained by the presence of hydrogen bridges between the nanoparticles. This means that a smooth fluidization is favored not only by low density agglomerates, but also by weak interparticle forces.
The results for the fluidization type, minimum fluidization velocity and bed expansion are shown in Fig. 3 and 4, and summarized in Table 2.
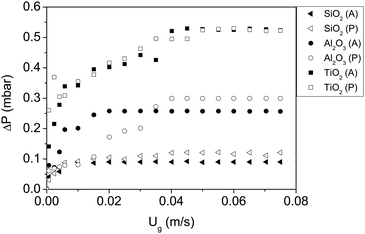 | ||
| Fig. 3 Pressure drop vs. gas velocity of all nanoparticles fluidized with dry nitrogen. P and A represent polar and apolar surfaces. | ||
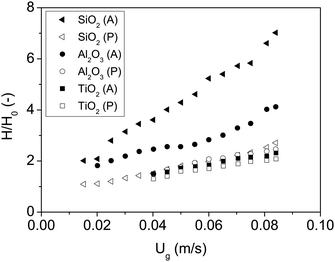 | ||
| Fig. 4 Bed expansion curves of all nanoparticles fluidized with dry nitrogen. P and A represent polar and apolar surfaces. | ||
| Material | Fluidization type | U mf [m s−1] | H/H0 at Ug = 0.05 [m s−1] |
|---|---|---|---|
| SiO2 (P) | ABF | 0.042 | 1.8 |
| SiO2 (A) | APF | 0.006 | 4.3 |
| Al2O3 (P) | ABF | 0.040 | 1.8 |
| Al2O3 (A) | ABF | 0.018 | 2.6 |
| TiO2 (P) | ABF | 0.050 | 1.6 |
| TiO2 (A) | ABF | 0.040 | 1.7 |
The first remarkable aspect is that polar particles present larger minimum fluidization velocity, and lower bed expansion than their apolar counterparts. The minimum fluidization velocity for small Reynolds and ρa ≫ ρf can be defined according to eqn (2):28
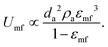 | (2) |
The information obtainable from the bed expansion is similar to that obtained from the minimum fluidization velocity. According to the Richardson and Zaki equation,32 in a non-bubbling fluidized bed Ug = Utεbn, where Ug is the gas velocity, Ut is the terminal velocity of a single particle (agglomerate), n ∼ 4.6, and εb is the bed voidage, which is related to the bed expansion. If Ut follows the Stokes law, then for a certain gas velocity and assuming that ρa ≫ ρf
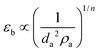 | (3) |
Since the fluidization gas is pure nitrogen, it is not expected to have substantial adsorption effects on the surface of the nanoparticles. Consequently, the differences in the results can be explained only by the formation of direct hydrogen bonds between the polar particles.
A simple calculation of the energy of cohesion between nanoparticles is shown in Table 3. The van der Waals interaction potential UvdW between two smooth spheres separated by a distance l is7
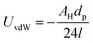 | (4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 J per mole, depending on the OH⋯O angle.22
000 J per mole, depending on the OH⋯O angle.22
| Core material | Interaction at the shortest scales | A H of the core material [J]33,34 | U vdW [J] eqn (4) |
|---|---|---|---|
| SiO2 (P) |
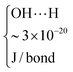
|
6.60 × 10−20 | ∼1 × 10−19 |
| Al2O3 (P) | 1.45 × 10−19 | ∼2 × 10−19 | |
| TiO2 (P) | 1.54 × 10−19 | ∼3 × 10−19 | |
| SiO2 (A) |
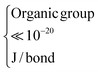
|
6.60 × 10−20 | ∼1 × 10−19 |
| Al2O3 (A) | 1.45 × 10−19 | ∼2 × 10−19 | |
| TiO2 (A) | 1.54 × 10−19 | ∼3 × 10−19 |
To estimate the effect of hydrogen bridges on different nanoparticles it is necessary to know the concentration of hydroxyl groups on the surface of each material and their configuration, since sometimes the hydroxyl groups form islands on the nanoparticle surface.20 As a first approach and simplifying as much as possible, the calculations listed in Table 3 show that the contribution of the hydrogen bonds to the total particle interaction is not negligible. A single hydrogen bond has an energy with the same order of magnitude as the potential energy between two nanoparticles.
These calculations also explain why polar and apolar SiO2 nanoparticles present the largest difference in the fluidization behavior. Due to the low Hamaker coefficient and small particle size of the SiO2 nanoparticles, the van der Waals potential between them is so low that the formation of only a few hydrogen bridges between them already provides a similar potential. On the other extreme, TiO2 nanoparticles show a minimum fluidization velocity and a bed expansion hardly affected by the presence of the hydrogen bonds. The van der Waals force between TiO2 particles is already large – both Hamaker coefficient and particle size are relatively large – so the formation of a few hydrogen bonds does not make a difference so appreciable like in the case of SiO2.
The second remarkable point is that the minimum fluidization velocity and the expansion curves are very similar for all polar nanoparticles, regardless of the material (Hamaker coefficient), particle size, and bulk density. Since the common factor in polar particles is just the presence of hydroxyl groups on the surface, the similarity in the results can be attributed to the formation of hydrogen bonds, which dramatically increases the interparticle attraction.
3.2 Fluidizing with nitrogen containing isopropanol vapour
To test the hypothesis that the formation of hydrogen bonds between polar nanoparticles dominates their interaction, the experiments were repeated fluidizing the powder with nitrogen containing ISP vapour. ISP is an amphiphilic compound with a hydroxyl group that can bind to the hydroxyl groups of the nanoparticles by means of hydrogen bridges. After bonding, the ISP molecules will expose the organic groups, decreasing the interaction between nanoparticles since the attraction between organic groups is weaker than that between hydroxyl groups.The minimum fluidization velocity and the expansion curves are shown in Fig. 5 and 6. The results are summarized in Table 4.
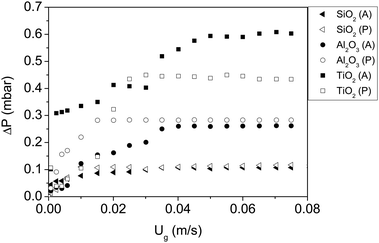 | ||
| Fig. 5 Pressure drop vs. gas velocity of all nanoparticles fluidized with dry nitrogen and ISP. P and A represent polar and apolar surfaces. | ||
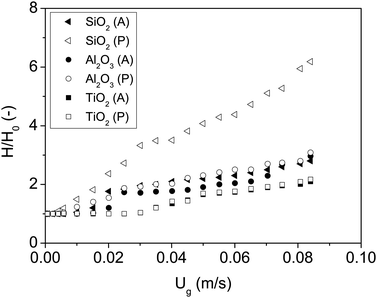 | ||
| Fig. 6 Bed expansion curves of all nanoparticles fluidized with dry nitrogen and ISP. P and A represent polar and apolar surfaces. | ||
| Material | Fluidization type | U mf [m s−1] | H/H0 at Ug = 0.05 m s−1 |
|---|---|---|---|
| SiO2 (P) | APF | 0.010 | 4.1 |
| SiO2 (A) | ABF | 0.035 | 2.2 |
| Al2O3 (P) | ABF | 0.015 | 2.3 |
| Al2O3 (A) | ABF | 0.037 | 1.9 |
| TiO2 (P) | ABF | 0.028 | 1.7 |
| TiO2 (A) | ABF | 0.047 | 1.7 |
It was shown in the previous sections that in the absence of ISP, all polar nanoparticles behaved similarly. However, the presence of ISP makes the particles behave differently: polar SiO2 has the smallest Umf and largest bed expansion, whereas TiO2 presents the largest Umf and smallest bed expansion. Moreover, polar nanoparticles in the presence of ISP behave almost identically to their apolar counterparts in the absence of ISP. The FTIR absorption spectrum reveals that the ISP molecules are adsorbed by hydroxyl groups of the nanoparticles, since the polar particles present peaks in the absorption spectrum that are consistent with the stretching vibration of the CH3 groups in the isopropanol molecule26 (Fig. 7). The same polar particles without ISP did not present these peaks (Fig. 2). Since the attraction between organic groups is weaker than that between hydroxyl groups, the attraction between polar nanoparticles decreases as a consequence of the ISP adsorption, as it can be inferred from the results.
 | ||
| Fig. 7 FTIR absorption spectra of the nanoparticles after being fluidized in the presence of ISP vapour. The peaks at 2976 cm−1 correspond to the stretching vibration of CH3 groups in the isopropanol molecule.26 | ||
In contrast, the presence of ISP molecules strongly increased the interaction between apolar nanoparticles, which can be derived from the increment in Umf and decrease of the bed expansion. Moreover, the presence of ISP makes the apolar nanoparticles behave almost like the polar nanoparticles in the absence of ISP. Under our experimental conditions, the adsorption of ISP adds a cohesive force between the apolar particles almost as strong as the force due to the hydrogen bonds that characterize the polar nanoparticles in the absence of ISP. An increase in the van der Waals force due to adsorption of species on the particle surface is in agreement with previously reported results.5,35,36 According to the results, the attraction between apolar nanoparticles is stronger than the attraction between their polar versions in the presence of ISP, which can be explained by a larger adsorption of ISP on the surface of apolar particles. This is in agreement with the results previously reported by Bettens et al.,37 who showed that methylated silica absorbs 50% more isopropanol than polar silica.
In this work we have qualitatively inferred the magnitude of the forces between nanoparticles in a fluidized bed from the minimum fluidization velocity and bed expansion. These two variables are related to the size of the agglomerates formed in the suspension.
Our results highlight the importance of the hydrogen bond interaction between nanoparticles in dense gas suspensions and the need of including this interaction in future modeling of the attraction between dry polar nanoparticles.
4 Conclusions
The formation of direct hydrogen bonds between the surface of dry polar nanoparticles strongly increases the interparticle force. The results reported have important implications in the theoretical estimation of the interaction between polar particles in the gas phase. If the formation of direct hydrogen bonds is not considered, the cohesion force between dry polar nanoparticles and the resulting agglomerate size will be strongly underestimated. Additionally, if the stronger attraction between polar particles in the presence of moisture is totally ascribed to capillary forces, this contribution will be overestimated.Acknowledgements
We would like to thank Andrea Fabre for commenting on the manuscript. Financial support from the European Research Council (Starting Grant) is gratefully acknowledged.References
- W. Yao, G. Guangsheng, W. Fei and W. Jun, Powder Technol., 2002, 124, 152–159 CrossRef.
- J. R. van Ommen, J. M. Valverde and R. Pfeffer, J. Nanopart. Res., 2012, 14, 737–766 CrossRef.
- J. Pauluhn, Toxicology, 2011, 279, 176–188 CrossRef CAS.
- G. Oberdorster, V. Stone and K. Donaldson, Nanotoxicology, 2007, 1, 2–25 CrossRef CAS.
- K. Rietema, Dynamics of Fine Powders, Springer, 1991 Search PubMed.
- J. P. K. Seville, C. D. Willett and P. C. Knight, Powder Technol., 2000, 113, 261–268 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, 2010 Search PubMed.
- H. Krupp, Adv. Colloid Interface Sci., 1967, 1, 111–239 CrossRef CAS.
- H. C. Hamaker, Physica, 1937, 4, 1058–1072 CrossRef CAS.
- N. A. Burnham, R. J. Colton and H. M. Pollock, Nanotechnology, 1993, 4, 64–80 CrossRef CAS.
- J. M. Valverde, M. A. S. Quintanilla, M. J. Espin and A. Castellanos, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2008, 77, 031301 CrossRef CAS.
- J. M. Valverde and A. Castellanos, Chem. Eng. J., 2008, 140, 296–304 CrossRef CAS.
- S. Salameh, J. Schneider, J. Laube, A. Alessandrini, P. Facci, J. W. Seo, L. C. Ciacchi and L. Madler, Langmuir, 2012, 28, 11457–11464 CrossRef CAS.
- L. F. Hakim, J. L. Portman, M. D. Casper and A. W. Weimer, Powder Technol., 2005, 160, 149–160 CrossRef CAS.
- S. Matsuda, H. Hatano, T. Muramoto and A. Tsutsumi, AIChE J., 2004, 50, 2763–2771 CrossRef CAS.
- X. S. Wang, F. Rahman and M. J. Rhodes, Chem. Eng. Sci., 2007, 62, 3455–3461 CrossRef CAS.
- S. Y. Wang, Y. R. He, H. L. Lu, J. X. Zheng, G. D. Liu and Y. L. Ding, Food Bioprod. Process., 2007, 85, 231–240 CrossRef.
- A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS.
- V. M. Gun'ko, V. I. Zarko, B. A. Chuikov, V. V. Dudnik, Y. G. Ptushinskii, E. F. Voronin, E. M. Pakhlov and A. A. Chuiko, Int. J. Mass Spectrom. Ion Processes, 1998, 172, 161–179 CrossRef CAS.
- M. Scholz and M. Kempf, Technical Bulletin Pigments, No. 12, Degussa AG technical report, 2001.
- Degussa, Technical Bulletin Pigments No. 11, Degussa AG technical report, 1997.
- C. Schilde, H. Nolte, C. Arlt and A. Kwade, Compos. Sci. Technol., 2010, 70, 657–663 CrossRef CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- B. Erdem, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4419–4430 CrossRef CAS.
- P. Larkin, Infrared and Raman Spectroscopy; Principles and Spectral Interpretation, Elsevier, 2011 Search PubMed.
- C. Zhu, Q. Yu, R. N. Dave and R. Pfeffer, AIChE J., 2005, 51, 426–439 CrossRef CAS.
- D. Kunii and O. Levenspiel, Fluidization Engineering, Butterworth-Heinemann, 1991 Search PubMed.
- S. C. Tsinontides and R. Jackson, J. Fluid Mech., 1993, 255, 237–274 CrossRef CAS.
- P. N. Loezos, P. Costamagna and S. Sundaresan, Chem. Eng. Sci., 2002, 57, 5123–5141 CrossRef CAS.
- C.-L. Lin, M.-Y. Wey and S.-D. You, Powder Technol., 2002, 126, 297–301 CrossRef CAS.
- J. F. Richardson and W. N. Zaki, Trans. Inst. Chem. Eng., 1954, 32, 35 CAS.
- H. D. Ackler, R. H. French and Y.-M. Chiang, J. Colloid Interface Sci., 1996, 179, 460–469 CrossRef CAS.
- L. Bergstrom, Adv. Colloid Interface Sci., 1997, 70, 125–169 CrossRef CAS.
- H.-Y. Xie, Powder Technol., 1997, 94, 99–108 CrossRef CAS.
- H. W. Piepers, E. J. E. Cottaar, A. H. M. Verkooijen and K. Rietema, Powder Technol., 1984, 37, 55–70 CrossRef CAS.
- B. Bettens, A. Verhoef, H. M. van Veen, C. Vandecasteele, J. Degreve and B. Van der Bruggen, J. Phys. Chem. C, 2010, 114, 9416–9423 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Owner Societies 2013 |