 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
KLaF4 nanocrystallisation in oxyfluoride glass-ceramics
A.
de Pablos-Martín
*ab,
F.
Muñoz
a,
G. C.
Mather
a,
C.
Patzig
b,
S.
Bhattacharyya
c,
J. R.
Jinschek
d,
Th.
Höche
be,
A.
Durán
a and
M. J.
Pascual
a
aInstituto de Cerámica y Vidrio (CSIC), C/ Kelsen 5, Campus de Cantoblanco, 28049 Madrid, Spain
bFraunhofer Institute for Mechanics of Materials IWM, Walter-Hülse-Str. 1, 06120 Halle, Germany. E-mail: araceli.de.pablos-martin@iwmh.fraunhofer.de
cDepartment of Condensed Matter Physics and Material Science, Tata Institute of Fundamental Research, Colaba, Mumbai 400005, India
dEurope Nanoport, FEI Company, Achtseweg Noord 5 Bldg, 5651 GG Eindhoven, The Netherlands
eLeibniz-Institute for Surface Modification, Permoserstraße 15, 04318 Leipzig, Germany
First published on 23rd September 2013
Abstract
Nanocrystallisation of the cubic and hexagonal polymorphs of KLaF4 in a 70SiO2–7Al2O3–16K2O–7LaF3 (mol%) glass has been achieved by heat treatment above the glass transition temperature. For treatment at 580 °C, only the cubic structure crystallises, with a maximum crystallite size of ~9 nm. At higher temperatures, crystallisation of the hexagonal structure also takes place. The crystallisation process has been analysed using several thermal and structural techniques and is revealed to occur from a constant number of nuclei. The formation of a viscous barrier which inhibits further crystal growth and limits the crystal size to the nanometric range is observed. The title materials doped with lanthanide ions may be good candidates for optical applications.
1. Introduction
In recent years, the production of glass-ceramics containing fluoride nano-crystals has played an increasingly significant role in the field of materials for luminescent applications.1,2 NaYF4 is known to be one of the most efficient hosts of rare-earth (RE) ions for near-infrared to visible up-conversion emission and has recently been investigated for its use in bio-imaging and tissue detection.3,4 The phase exists in high-temperature cubic and low-temperature hexagonal polymorphs.5 Both crystal symmetries have been investigated for exhibiting interesting luminescent properties when lanthanide ions are incorporated in their crystalline structures. NaYF4 particles of different morphologies have been prepared by using several solution methods, such as hydrothermal6 and solvothermal techniques.7 Wang et al.8 synthesised both polymorphs separately by using the solvothermal method and reported that the up-conversion emission in Tm/Yb-doped NaYF4 microcrystals is more efficient in the hexagonal structure than in the cubic phase, due to the incorporation of RE ions in multiple sites of the former. Chai et al.9 prepared hexagonal NaYF4 nano-particles co-doped with Er3+/Yb3+ and Tm3+/Yb3+ cations by using in situ polymerisation. NaYF4 crystallisation is also reported to take place in glass-ceramics obtained either by using the sol-gel method or by melting and heat treatment. Yanes et al.10 prepared glass-ceramics by a sol-gel process, in which cubic α-NaYF4 nano-crystals co-doped with Er3+/Yb3+ were formed. Liu et al.11 reported the crystallisation mechanism of an Nd3+-doped oxyfluoride glass, prepared from melting and subsequent heat treatment, where ca. 30 nm cubic NaYF4 crystals precipitated and Nd3+ ions act as the nucleation agent. Zhao et al.12 also achieved cubic NaYF4 nano-glass-ceramics by melting and heat treatment of a borosilicate glass and observed an important enhancement of the up-conversion luminescence of Er3+/Yb3+ ions in the glass-ceramic in comparison with the parent glass.The enhancement of the luminescent properties of lanthanide ions when incorporated in NaYF4 crystals and the multisite nature of this phase have motivated the preparation of further RE-doped glass-ceramics containing fluoride nanocrystals in an AREF4-type host (A = alkali metal). Herrmann et al.,13 for example, recently developed oxyfluoride glass-ceramics in which hexagonal and cubic NaGdF4 nano-crystals precipitate. Sroda14 reported on the effect of the addition of Er2O3 in a glass in the system SiO2–Al2O3–Na2O–Al2F6–LaF3, where NaLaF4 crystallises.
In previous work,15 we described the nano-crystallisation mechanism of hexagonal NaLaF4 in a glass of composition 70SiO2–7Al2O3–8K2O–8Na2O–7LaF3 (mol%), which is found to result from enrichment of silica at the interphase between the crystals and the glassy matrix. The compositional gradient leads to the formation of a diffusional barrier which inhibits further crystal growth.16 In subsequent studies, the characterisation of similar glass-ceramics containing NaLaF4 and LaF3 nano-crystals doped with Tm3+ was reported,17,18 and the partial distribution of Tm3+ between the glassy matrix and crystals was confirmed. Another example of the crystallisation of AREF4 phases includes RE-doped cubic KLaF4 nano-particles, obtained by using solution methods.19,20 Filatova et al.21 reported the phase diagram of the KF–LaF3 system, in which KLaF4 presents both a cubic fluorite structure, α-KLaF4, and a hexagonal low-temperature form, β-KLaF4,22 analogous to the NaYF4 polymorphs. Alternatively, preparation of the hexagonal phase by fusing the fluoride components, first reported by Zachariasen,22 is becoming a popular synthesis method.23 A phonon energy of 262 cm−1 has been reported for hexagonal KLaF4.23 Tyagi et al.19 suggested that the advantageous lower phonon energy of KLaF4 in comparison with NaYF4 (360 cm−1) arises from the greater radii of the alkali-metal ion, whereas the greater size of the rare-earth ion in KLaF4 as compared to NaYF4 imparts higher stability.24 However, to the best of our knowledge, glass-ceramics containing KLaF4 have not been reported until now.
This work is focussed on the preparation of oxy-fluoride glass-ceramics containing KLaF4 nano-crystals and the description of the crystallisation mechanism based on both our previous studies15,16 and numerous new experimental results. The potential of these glass-ceramics as hosts for rare-earth ions for optical applications is discussed.
2. Experimental
A glass of nominal composition 70SiO2–7Al2O3–16K2O–7LaF3 (mol%) was prepared by melting reagent grade SiO2 sand (Saint Gobain, 99.6%), Al2O3 (Panreac, 99.5%), K2CO3 (Panreac, 99.5%) and LaF3 (Panreac, 99%) in an electric furnace. The batches were first calcined in covered platinum crucibles at 1200 °C and then melted for 1 h at 1600 °C under the same conditions. The melts were quenched in air onto a brass mould and then annealed above the glass transition temperature (Tg). The glasses were analysed by using X-ray Fluorescence Spectroscopy (XRF) with a Panalytical spectrometer. All oxides were determined employing the melting method with Li2B4O7, whereas elemental fluorine analysis was performed on pressed pellets of powdered glass (8 g) in order to avoid fluorine volatilization.T g was determined by using dilatometry in a Netzsch Gerätebau dilatometer, model 402 EP, with a 10 K min−1 heating rate in air; the estimated error of Tg is ±1 °C.
The viscosity–temperature curve of the glass was determined by combining different series of data: Tg (obtained from the DSC curve at 10 K min−1), corresponding to a viscosity of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) η = 13.5 (η in dPa s), and viscosity measurements obtained from both a high-temperature viscometer and a hot-stage microscope. The measurement of glass viscosity in the range 105–101 dPa s was conducted by the rotation procedure according to International Standard ISO 7884-2 with a high-temperature Haake viscometer equipped with a ME 1700 cylindrical Searle-type sensor at rotation speeds of 1–30 rpm for 10 min. Three measurements were taken at three different rotation speeds for each measured temperature.
η = 13.5 (η in dPa s), and viscosity measurements obtained from both a high-temperature viscometer and a hot-stage microscope. The measurement of glass viscosity in the range 105–101 dPa s was conducted by the rotation procedure according to International Standard ISO 7884-2 with a high-temperature Haake viscometer equipped with a ME 1700 cylindrical Searle-type sensor at rotation speeds of 1–30 rpm for 10 min. Three measurements were taken at three different rotation speeds for each measured temperature.
Hot-stage microscopy (HSM) was performed with a Leica-EM201 microscope with image analysis for determination of the viscosity in air at a rate of 5 K min−1. The samples were initially cold pressed to form bodies of 3 mm in both height and diameter from a glass powder with a particle size <60 μm. The temperature was measured with a Pt/Rh (6130) thermocouple placed under and in contact with the alumina support. The temperatures corresponding to the characteristic viscosity points, following Scholze's definition,25 were obtained from photographs taken in the following sequence: first shrinkage (logη = 9.1 ± 0.1), maximum shrinkage (logη = 7.8 ± 0.1), softening (logη = 6.3 ± 0.1), half ball (logη = 4.1 ± 0.1) and flow point (logη = 3.4 ± 0.1); the error in temperature is ±0.2 °C and η is in dPa s.
Glass-ceramics were obtained by controlled crystallisation of cubic samples that were heat treated at temperatures close to Tg for between 1 and 200 h. Kinetic studies were also performed in order to determine the influence of the treatment time at temperatures close to Tg.
Powder X-ray diffraction (XRD) was carried out with an X'Pert PRO Panalytical diffractometer equipped with an X'Celerator detector. The patterns were collected with monochromatic Cu Kα radiation (1.5406 Å) over the angular range 10 ≤ 2θ ≤ 60° in a step size of 0.0167° and a fixed counting time of 1.63 s per step. The size of crystallites was estimated using the Scherrer equation (eqn (1)) using the 2θ ≈ 43° diffraction peak:
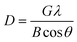 | (1) |
19F Magic Angle Spinning Nuclear Magnetic Resonance (MAS-NMR) spectra of the parent glass and those glass-ceramics obtained after annealing at 580 °C, were collected employing a Bruker AV 400 WB spectrometer operating at 376 MHz (9.4 T) and using a 2.5 mm probe. The pulse length was 2 μs, with a delay time of 30 s. A total number of 256 scans were accumulated with a spinning rate of 25 kHz. Solid CaF2 was used as secondary reference with a chemical shift of −0.108 ppm with respect to a CFCl3 reference.
Samples of glass and glass-ceramic for analysis using transmission electron microscopy (TEM) were prepared by cutting into slices, plane-parallel grinding, and dimpling to a residual thickness of ca. 15–20 μm. The samples were then subjected to double-sided, ion-beam milling using low-energy (2.5 keV) Ar+ ions under a small angle of incidence (6°) until the central part of the samples became electron transparent (precision ion-polishing system, PIPS, Gatan Company). The non-conducting samples were coated over selected areas with carbon using a special coating mask27 prior to TEM investigation in order to reduce charging effects resulting from interaction with the electron beam. All the samples were found to degrade in the electron beam, so an acceleration voltage of just 75–80 kV was used in order to minimise sample damage. TEM analyses of the base glass and the glass-ceramics obtained by heat treatment at 580 °C for 60 h were performed with a Hitachi H-8100 transmission electron microscope operated at 75 kV. High-resolution images of the precipitated crystals were registered with a FEI Titan3 80-300 electron microscope (FEI Company) operated at 80 kV at the Centre for Electron Nanoscopy (Cen), Technical University of Denmark (DTU).
Scanning transmission electron microscopy (STEM) can present advantages over TEM for the analysis of delicate samples because the electron dose is lower in STEM, which can be crucial for electron-sensitive samples, and illumination of the sample is performed with a parallel electron beam rather than with a fine-focused probe. Moreover, the STEM data can be inherently combined with spatially resolved analytics in terms of a fine spot used for both imaging and energy-dispersive X-ray spectroscopy (EDXS).28 The microstructure of the glass-ceramic obtained by annealing at 600 °C for 60 h was characterised by STEM using a high-angle annular dark-field (HAADF) detector (Fischione model 3000; camera length, 145 mm), using a Titan3 80-300 electron microscope operating at 80 kV. Elemental mappings of different elements were obtained in order to gain information about the elemental distribution with high spatial resolution on combination of STEM data with energy dispersive X-ray spectroscopy (EDXS), obtained by means of a SuperX-EDXS detector (FEI Company) consisting of four silicon drift detectors offering a maximum collection angle of 0.9 sr above the sample surface. Elemental mappings were obtained by evaluating the integrated peak intensity of either the Kα- (O, F, K, Al, Si) or Lα- (La) edges of the respective elements, with an automatic routine provided by the commercially available software Esprit (Bruker Company).
Differential scanning calorimetry (DSC) measurements were performed with a Setaram instrument, Model Setsys Evolution 16/18, using powdered Al2O3 as an inert reference material. A quantity of glass of 100–150 mg with a particle size in the range 1–1.25 mm was used in order to reproduce conditions similar to the bulk glass.
3. Results and discussion
3.1. Chemical analysis and dilatometry
The synthesized oxyfluoride glass was transparent. The X-ray diffractogram of the parent glass (not shown) did not reveal the presence of any crystalline phases.The chemical analysis shown in Table 1 provides the weight percentage (wt%) of the oxide form of each element, together with the fluorine content. Fluorine loss during the melting procedure was calculated as 31 wt% by subtracting the nominal and the analysed fluorine quantities, which is a value lower than similar glasses15,16 in which losses around 40 wt% were calculated.
| Components | Nominal wt% | Analysed wt% | Nominal mol% | Analysed mol% |
|---|---|---|---|---|
| SiO2 | 53.94 | 55.01 | 70 | 72.79 |
| Al2O3 | 9.15 | 7.64 | 7 | 5.97 |
| K2O | 19.32 | 17.67 | 16 | 14.88 |
| LaF3 | 17.58 | 11.75 | 7 | 4.77 |
| La2O3 | — | 6.52 | — | 1.59 |
| F | 5.11 | 3.50 | 21 | 12.79 |
The glass transition temperature, Tg, of the parent glass was determined by dilatometry to be 552 ± 2 °C, and the dilatometric softening point, Td, was 629 ± 2 °C.
3.2. Viscometry
Fig. 1 shows the viscosity–temperature curve of the glass determined from different techniques. The temperature dependence of viscosity was fitted using empirical Vogel–Fulcher–Tammann (VFT)29–31 and Avramov32 equations, following the same procedures as adopted in ref. 15 and 16. Both equations fit satisfactorily to the experimental data such that their curves are coincidental (Fig. 1). It is interesting to compare the studied oxyfluoride glass and that analysed in previous work,15 in which 50 mol% of the total K2O content in the studied glass is substituted by Na2O, giving a mixed alkali composition of 70SiO2–7Al2O3–8K2O–8Na2O–7LaF3 mol%.15 The VFT/Avramov fits of the corresponding viscosity–temperature curve of this mixed-alkali glass were reported in ref. 15 and are also shown in Fig. 1. The mixed-alkali glass presents a lower viscosity than that of the glass in the present work in the studied temperature range, mainly due to the mixed-alkali effect.33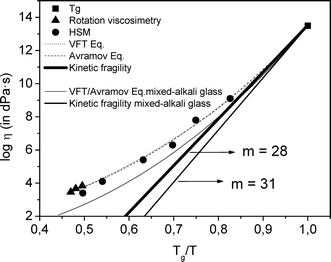 | ||
| Fig. 1 Viscosity–temperature curve of the glass, determined by using Tg point, hot-stage microscopy (HSM) and rotation methods. Curves were fitted according with the Vogel–Fulcher–Tammann (VFT) and Avramov–Milchev (AM) equations. Kinetic fragility is also plotted. | ||
Angell34 has described the strong/fragile behaviour of glasses by the m parameter, which is higher for more fragile glasses and is also a measure of the deviation from Arrhenian temperature dependence. From Angell's plot, the kinetic fragility parameter m can be calculated.16 From Fig. 1, kinetic fragility of the studied glass of m = 28 is extracted, while m = 31 in the mixed-alkali glass. That is, the viscosity of the non-mixed alkali glass is less sensitive to temperature changes.
Since the viscous flow is a thermally activated process, the temperature dependence of viscosity can also be described in terms of the activation energy of the viscous flow.30 The activation energy of the viscous flow of the studied glass was calculated in the whole temperature range (plot not shown) and found to be higher but with a more moderate variation of the activation energy with temperature than that of the mixed-alkali glass,15 in agreement with its lower fragility value.
3.3. X-ray diffraction
The parent glass was heat treated at 580 °C from 1 to 340 h (14 days), after which, transparent glass-ceramics were obtained, even for the longest treatment times. The XRD patterns of the glass-ceramics are shown in Fig. 2. Diffraction peaks are visible after 3 h at 580 °C and correspond with those of cubic α-KLaF4 (JCPDS 075-2020). The diffraction peaks become more intense with increasing time of treatment, indicating an increase in crystalline fraction. It is important to highlight the influence of the glass composition in the resulting crystalline phase. The crystallisation of KLaF4 reported in this work does not take place on heat treatment of the mixed alkali glass reported in ref. 15, in which NaLaF4 crystals are formed instead.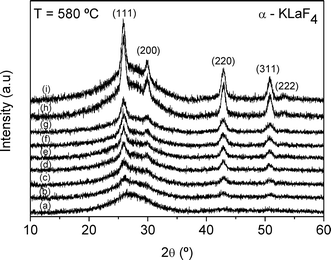 | ||
| Fig. 2 XRD patterns of the parent glass heat treated at 580 °C for (a) 1 h, (b) 3 h, (c) 5 h, (d) 10 h, (e) 24 h, (f) 95 h, (g) 150 h, (h) 217 h and (i) 340 h. | ||
The size of the α-KLaF4 crystallites as a function of time of treatment at 580 °C, Fig. 3, varies little around a mean value of ~9 nm, although a slight increase in size is observed in the glass-ceramics obtained after 217 and 340 h. Nevertheless, it is apparent that the crystal growth velocity is very slow at this temperature, in accordance with the corresponding high viscosity value of logη = 12 (η in dPa s), Fig. 1. The intensity of the diffraction peaks increases progressively until 217 h of treatment. Thus, it can be assumed that the crystalline fraction increases with the time of treatment, whereas the crystal size remains practically constant (Fig. 3), indicating an efficient crystallisation mechanism which produces an increasing quantity of nano-crystals of similar size. Such behaviour is likely to be attributable to the existence of a viscous layer around the crystals which hinders further crystal growth.15 A narrow crystal-size distribution which is almost independent of the duration of heat treatment is achieved, as previously reported for similar glass-ceramics containing nanocrystals.15,16,35
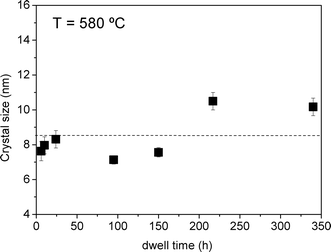 | ||
| Fig. 3 Crystal size as a function of time of treatment at 580 °C. | ||
Treatments at higher temperatures, 600 and 660 °C for 60 h, lead to the additional crystallisation of β-KLaF4 with hexagonal symmetry (JCPDS 075-1927, Fig. 4), with retention of transparency. Although the intensity and the narrowness of the peaks of both phases increase from 600 °C to 660 °C, it is more significant for the β-KLaF4 peaks, indicating that increasing temperature favours crystallisation of the hexagonal phase. Liu et al.11 reported the crystallisation of cubic NaYF4 nano-crystals as the only phase in a glass-ceramic in the system SiO2–Al2O3–Na2O–YF3–NdF3. According to the NaYF4 phase diagram proposed by Thoma et al.,5 the hexagonal β-NaYF4 is the thermodynamically stable phase at low temperatures and the cubic α-NaYF4 is the high-temperature modification. Crystallisation of NaYF4 takes place in a metastable glassy system far from the equilibrium state such that the cubic α-NaYF4 phase is obtained as the kinetically stable product.11 However, analogous to our results, the crystallization of cubic NaGdF4 nano-crystals in an oxyfluoride glass matrix occurs at lower temperatures,13 up to 600 °C, while at higher temperatures, above 650 °C, X-ray diffraction peaks of the hexagonal NaGdF4 polymorph are detected, whereas the peaks corresponding to the cubic phase decrease in intensity. In other compositions of the same work,13 the hexagonal phase of NaGdF4 crystallises as the only phase. It is proposed that a higher concentration of Na2O results in crystallisation of the hexagonal phase, whereas a higher concentration of dopant, such as Gd2O3, favours precipitation of the cubic phase.13
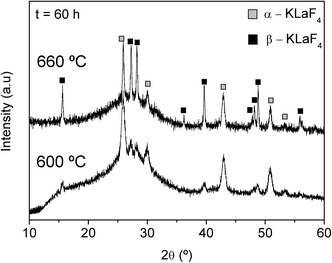 | ||
| Fig. 4 XRD patterns of the parent glass heat treated at 600 °C and at 660 °C for 60 h. | ||
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), β-KLaF4 (P6
m), β-KLaF4 (P6![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) m), with respective internal standards of NaF (Fmm) and the anatase form of TiO2 (P42/mnm), were refined by Rietveld analysis based on published structural data.36,37 The thermal vibration parameters of the phases were kept constant in order to avoid correlation with the background or other parameters.38 The observed diffraction patterns and the difference patterns between observed and calculated data on termination of refinement are shown in Fig. 5a and b for the glass ceramics treated at 580 °C for 150 h and 600 °C for 60 h, respectively. The corresponding structural parameters and reliability factors indicating the quality of the refinement are listed in Tables 2 and 3.
m), with respective internal standards of NaF (Fmm) and the anatase form of TiO2 (P42/mnm), were refined by Rietveld analysis based on published structural data.36,37 The thermal vibration parameters of the phases were kept constant in order to avoid correlation with the background or other parameters.38 The observed diffraction patterns and the difference patterns between observed and calculated data on termination of refinement are shown in Fig. 5a and b for the glass ceramics treated at 580 °C for 150 h and 600 °C for 60 h, respectively. The corresponding structural parameters and reliability factors indicating the quality of the refinement are listed in Tables 2 and 3.
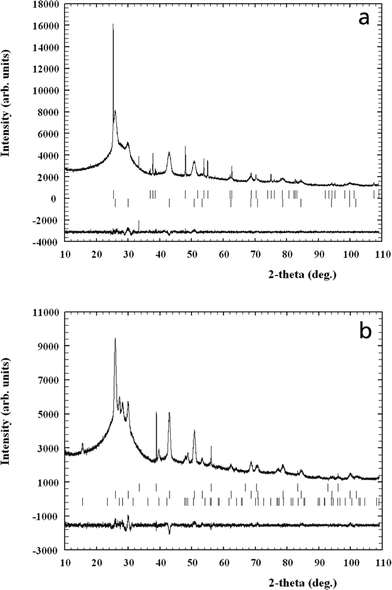 | ||
| Fig. 5 (a) Observed and difference X-ray powder diffraction profiles of the glass-ceramic obtained at 580 °C, 150 h, with 3 wt% TiO2 as an internal standard. The Bragg peaks of TiO2 and cubic KLaF4 are indicated by top and bottom vertical bars, respectively. (b) Observed and difference X-ray powder diffraction profiles of the glass-ceramic obtained at 600 °C, 60 h, with 0.8 wt% NaF as an internal standard. The Bragg peaks of NaF and cubic and hexagonal KLaF4 are indicated by top, middle and bottom vertical bars, respectively. | ||
m; a = 5.9552(7) Å; Rp = 2.11; Rwp = 2.93; χ2 = 1.95a
| Atom | Site | x/a | y/b | c/z | Occupation |
|---|---|---|---|---|---|
| a Thermal vibration parameters were constrained to Biso = 0.8. | |||||
| K(1) | 4a | 0 | 0 | 0 | 0.5 |
| La(1) | 4a | 0 | 0 | 0 | 0.5 |
| F(1) | 8c | 0.25 | 0.25 | 0.25 | 1 |
m; a = 6.550(1) Å; c = 3.805(1); Rp = 2.17; Rwp = 2.81; χ2 = 1.80a
| Atom | Site | x/a | y/b | c/z | Occupation |
|---|---|---|---|---|---|
| a Thermal vibration parameters were constrained to Biso = 0.8 | |||||
| K(1) | 2d | 0.3333 | 0.6667 | 0.5 | 0.75 |
| La(1) | 2d | 0.3333 | 0.6667 | 0.5 | 0.25 |
| La(2) | 1a | 0 | 0 | 0 | 1 |
| F(1) | 3f | 0.592(5) | 0 | 0 | 1 |
| F(2) | 3g | 0.167(4) | 0 | 0.5 | 1 |
In the case of treatment at 580 °C for 150 h, only the α form of KLaF4 crystallises; we note the presence of a small unidentified reflection at 33.3° 2θ, which is considered to be an artefact due to the fact that this peak does not appear at higher treatment temperature (Fig. 4) and is much sharper in profile than the identified crystallised phases. The corresponding crystallite size of α-KLaF4 calculated from the refinement is 9 nm, in very good agreement with that presented in Fig. 3. On treatment at 600 °C for 60 h, both α and β polymorphs are present. The corresponding calculated crystal sizes in this glass-ceramic are ~20 nm and ~15 nm for the α and β nanocrystals, respectively. Thus, the α polymorph grows with temperature from 9 nm at 580 °C (Fig. 3) to 20 nm at 600 °C, while the additional β polymorph crystallises.
The weight fractions of α and β-KLaF4 obtained by refinement were corrected for the presence of the amorphous phase from the X-ray and real weight fractions of the internal standards with the same procedure as outlined in previous work.15,16 Absorption contrast effects (microabsorption) of phase mixtures with different linear absorption coefficients may influence the X-ray diffraction intensities and the results of the quantitative analysis. Brindley39 classified the absorption effects in mixed powders on the basis of the value of μD where μ is the linear absorption coefficient and D is the particle size. In the present case, the KLaF4 nanocrystals and internal standards may be classified as fine powders (μD < 0.01) on the basis of their weighted absorption coefficients and particle sizes such that no microabsorption correction needs to be applied. The final corrected weight fraction of α-KLaF4 in the glass ceramic treated at 580 °C for 150 h was 5.8 wt%, while the glass-ceramic treated at 600 °C for 60 h presents the same quantity of the α polymorph, 5.8 wt%, and 2.1 wt% of the β phase. Thus, with increasing temperature, crystallisation of the hexagonal phase takes place whereas the quantity of cubic phase remains constant. This is further clarified by the results of TEM and EDX analyses presented in later sections.
3.4. 19F Nuclear magnetic resonance
Fig. 6 shows the 19F MAS NMR spectra of the glass and glass-ceramics obtained after 60, 72 and 150 h of heat treatment at 580 °C. The spectrum of the glass exhibits three intense resonances, at ca. −18, −138 and −176 ppm, the first being a very broad undefined band likely comprising several resonances. According to previous results reported by Muñoz et al.40 concerning the nano-crystallization of LaF3 and NaLaF4 phases, this broad band may be attributable to different local environments of fluorine, K–F–La bonds in the present case, which act as precursor nuclei of the fluoride crystals that grow after heat treatment. All three resonances show typical broadening of nuclei in an amorphous environment. The resonance at −138 ppm may be attributed to F–K bonds, such as from KF,41 while the third resonance, centred at −176 ppm, is assigned to fluorine atoms in Al–F–Al bonds, as in AlF3.41 An estimation of the ratio between the areas of the different resonances has been carried out through deconvolution of the main resonances in the spectra by using DMFIT software.42 The ratio between the area of the band at −18 ppm (K–F–La bonds) and that of the band at −176 ppm (Al–F–Al) is around 3, while that between the former and the band at −138 ppm (F–K) is around 7. This indicates the predominance of fluorine in K–F–La environments in this glass.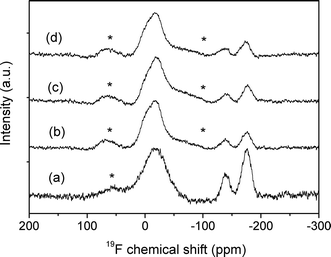 | ||
| Fig. 6 19F NMR spectra of (a) the glass and glass-ceramics obtained at 580 °C after (b) 60, (c) 72 and (d) 150 h. ( * denotes spinning side bands.) | ||
After heat treatment, there is a clear decrease of the intensity of the bands at −176 ppm (Al–F–Al) and at −138 ppm (F–K) with respect to the resonance at −18 ppm, while their intensities do not change significantly by comparing the three glass-ceramics. The fact that the resonances assigned to the KLaF4 crystals appeared at a chemical shift close to the resonance centred at ca. −18 ppm could indicate that crystals are formed from nuclei present in the glass. However, as seen in the decrease of the NMR intensities of the Al–F–Al and F–K bonds in the glass-ceramic samples, fluorine should also diffuse from the Al–F–Al and K–F bonds to KLaF4 crystals.
3.5 Transmission electron microscopy
Fig. 7a shows a TEM bright-field micrograph of the homogeneous parent glass in which no liquid–liquid phase separation occurs, similar to the glass reported in ref. 15, but in contrast with other oxyfluoride glasses in which phase separation is observed.16 The glass-ceramic obtained after annealing at 580 °C for 60 h, Fig. 7b, consists of nano-crystals of ca. 20 nm in diameter, which are homogenously distributed and with a limited size, in accordance with the narrow size distribution observed in Fig. 3. The crystal size in the TEM micrograph corresponds to the particle size, which is different to that of a single crystalline domain calculated from XRD analysis (9 nm, Fig. 3). Thus, the crystals observed in the micrograph in Fig. 7b would seem to correspond to crystalline domains of 9 nm forming spherical nano-particles of ca. 20 nm in diameter. Based on previous studies, the remaining volume is expected to be filled with silica or even alumina,18 as discussed later. | ||
| Fig. 7 TEM micrographs of the base glass (a) and glass-ceramic obtained at 580 °C for 60 h (b). | ||
Fig. 8a shows the STEM micrograph of the glass-ceramic obtained at higher temperature, 600 °C for 60 h, where crystals of different sizes are observed. The biggest crystals have a diameter of between 20 and 25 nm and the smallest of ~10 nm. Fig. 8b shows a HRTEM micrograph of a 20 nm diameter crystal, in which crystalline planes may be attributed to both α- and β-KLaF4 structures. Thus, the larger crystals observed in Fig. 8a exhibit both α- and β-KLaF4 structures. Another 20 nm crystal together with a smaller one of 10 nm diameter is shown in Fig. 8c. The latter consists of only the α-KLaF4 phase. It can, thus, be concluded that at lower temperatures (580 °C, 150 h), α-KLaF4 nano-crystals precipitate (Fig. 7b), whereas at higher temperatures (600 °C, 60 h), β-KLaF4 domains are formed close to the α-KLaF4 nano-crystals (Fig. 8b). Additionally, α-KLaF4 nano-crystals of smaller crystal size precipitate from the remaining fluorine in the glassy matrix. This is consistent with a transformation of the α polymorph to the β phase in a quantity which is compensated by the additional crystallisation of smaller α-KLaF4 crystals, such that the amount of the α phase, given as 5.8 wt% by Rietveld refinement, remains constant in both glass-ceramics.
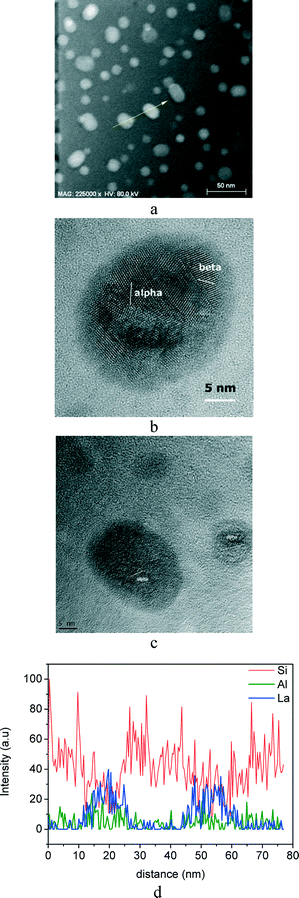 | ||
| Fig. 8 STEM micrographs of the glass-ceramic obtained at 600 °C after 60 h (a) and the corresponding HRTEM micrographs (b, c); (d) compositional analysis of an EDXS line scan across two nano-crystals, indicated with a yellow arrow in (a). | ||
Cubic α-KLaF4 is the high-temperature, metastable phase whereas the hexagonal (β-) form is the thermodynamically stable polymorph.19 This behaviour is, thus, consistent with Ostwald's rule of stages, according to which the metastable phase is preferably formed first,43,44 and subsequently transforms to the thermodynamically stable phase. In the present glass, the viscosity decreases significantly at higher temperatures from log η = 12 at 580 °C to log η = 11.5 and log η = 9.4 at 600 and 660 °C, respectively (Fig. 1). Thus, at high temperatures, the diffusional barrier is less efficient for preventing crystal growth and the crystal growth rate increases.16 This situation could favour the crystallisation of the thermodynamically stable hexagonal KLaF4 phase, whereas at lower temperatures (580 °C), crystallisation of cubic, metastable α-KLaF4 as the only phase is favoured under the effect of the diffusional barrier.
EDXS was employed in order to further study the crystallisation mechanism; an EDXS line scan across two 20 nm diameter nano-crystals, indicated in Fig. 8a by the arrow, is shown in Fig. 8d. For the sake of clarity, only Si, Al and La intensities are represented. The relatively poor signal-to-noise ratio is due to the fact that beam intensity was adjusted to accommodate the radiation sensitivity of the glass-ceramics, ensuring that detection was independent of sample changes occurring on measurement. The intensity of the La signal clearly increases in the crystalline areas containing KLaF4 nano-crystals, but is hardly detectable in the surrounding area. The intensity of the Si signal, on the other hand, decreases in the crystalline areas, indicating an enrichment of Si in the periphery of the crystals, which could constitute the above-mentioned silica diffusional barrier. The intensity of the Al signal in Fig. 8d is almost constant across the line scan, although a slight increase in the periphery of the crystalline area can be observed. Bhattacharyya et al.45 reported an alumina enrichment around ZrTiO4 precipitates of ~4 nm in a lithium aluminosilicate glass, which provides suitable preconditions for the nucleation of a secondary high-quartz phase. The formation of an alumina shell in the studied glass-ceramic cannot, therefore, be excluded. However, the Al2O3 content in the studied glass composition is only 7 mol%, and it is thus difficult to discern enrichment around the crystals arising from the alumina remaining in the glassy matrix.
3.6. Differential scanning calorimetry
DSC curves of the studied glass employing heating rates from 5 to 40 K min−1 are shown in Fig. 9. The maximum of the main crystallisation peak, Tp, shifts to higher temperatures with an increasing heating rate, from 635 °C at 5 K min−1 to 667 °C at 40 K min−1. Heat treatment of the parent glass at 652 °C for 1 h, which corresponds to the Tp at 10 K min−1, results in the crystallisation of cubic α-KLaF4 and a small contribution of the β-KLaF4 phase (XRD diffractogram not shown).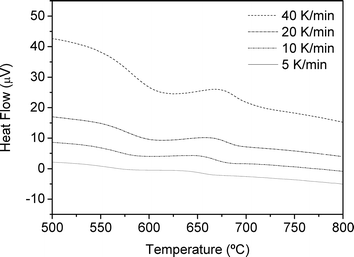 | ||
| Fig. 9 DSC curves of the glass recorded with heating rates in the range 5–40 K min−1. | ||
Following the same procedure as that adopted in previous work,16,46 calculation of the Avrami exponent n was performed from the Ozawa equation.47 The plot used to calculate n is shown in Fig. 10 and gives an average value of n = 1.29 (Table 4).
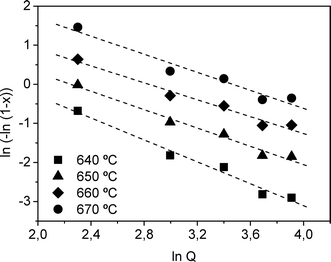 | ||
| Fig. 10 Ozawa plot constructed from DSC data from 640 to 670 °C for the determination of the Avrami exponent n. | ||
| T (°C) | 640 | 650 | 660 | 670 |
|---|---|---|---|---|
| n ± 0.1 | 1.4 | 1.2 | 1.1 | 1.2 |
The activation energy for crystallization was calculated applying the Kissinger, Takhor and Augis-Bennett (KTAB) equations,48Fig. 11a.
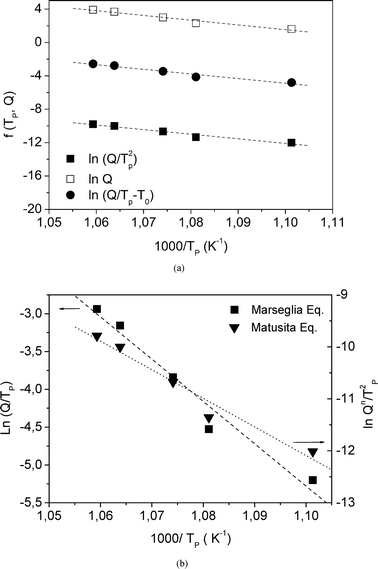 | ||
| Fig. 11 (a) ■ Kissinger, □ Takhor and ● Augis-Bennett plots constructed from the DSC data; (b) Marseglia and Matusita plots. Q is the heating rate and Tp is the DSC crystallisation peak. | ||
The Matusita equation can also be applied49 (Fig. 11b) in order to obtain the parameter m (dimensionality of crystal growth). In the present work, E/n is first obtained from the Marseglia equation46 (Fig. 11b). The n value determined from the Ozawa equation allows the activation energy E from the Marseglia plot to be calculated, giving a value of 450 kJ mol−1. The activation energy determined from the KTAB equations is equal to that obtained via the Marseglia relation, with an average value of 450 kJ mol−1 (Table 5). The value of n obtained from the Ozawa equation then permits calculation of the activation energy from a plot of the Matusita relation (Fig. 11b), from which m can also be determined. The m value, 0.98, approaches the average obtained for n of 1.3. According to Donald,50 these m and n values are attributable to a bulk crystallisation mechanism with a constant number of nuclei (i.e., a well-nucleated sample in which the number of nuclei is independent of the heating rate) involving two growth dimensions, with a crystal size growth rate of 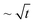 (diffusion controlled). In this type of mechanism, similar to that found in other oxyfluoride glasses,16m = n = 1, and use of the KTAB equations is appropriate. The 19F NMR results, in which K–La–F bonds were detected in the parent glass as precursors of the KLaF4 nano-crystals, support this mechanism model.
(diffusion controlled). In this type of mechanism, similar to that found in other oxyfluoride glasses,16m = n = 1, and use of the KTAB equations is appropriate. The 19F NMR results, in which K–La–F bonds were detected in the parent glass as precursors of the KLaF4 nano-crystals, support this mechanism model.
| Activation energy (kJ mol−1) | |||
|---|---|---|---|
| ln Q Takhor | ln(Q/Tp2) Kissinger | ln(Q/(Tp − T0)) Augis-Bennett | ln(Q/Tp) Marseglia |
| 458 ± 9 | 442 ± 9 | 446 ± 9 | 450 ± 9 |
From the series of presented results, it can be stated that α-KLaF4 crystallises with a very narrow crystallite size distribution of 9 nm (calculated from XRD results) at 580 °C, which remains constant with time due to the effect of a diffusional barrier which limits the crystal growth. At higher temperatures, β-KLaF4 crystallizes from the α-KLaF4 nano-crystals and, additionally, α-KLaF4 precipitates from the fluorine remaining in the glassy matrix. The periphery of the precipitated crystals was confirmed to be enriched in silica, although the additional formation of an alumina shell cannot be excluded. Both shells would constitute a diffusional barrier which limits crystal growth. The presence of K–La–F bonds in the parent glass, confirmed by 19F NMR, gives rise to the α-KLaF4 nano-crystals after heat treatment. Moreover, a diffusional process from K–F and Al–F–Al to the K–F–La environments takes place. DSC analysis confirms that the crystallization mechanism is bulk in character and takes place from an initially nucleated glass in a diffusion-controlled process, in agreement with the results of 19F NMR.
4. Conclusions
Transparent oxyfluoride glass-ceramics containing KLaF4 nano-crystals were obtained from thermal treatments of a 70SiO2–7Al2O3–16K2O–7LaF3 (mol%) glass at temperatures from Tg + 30 to Tg + 100 K. The alkali present in the glass composition has an important influence on the viscosity and kinetic fragility of the glass. The studied glass has been compared with an analogous mixed-alkali glass of composition 70SiO2–7Al2O3–8K2O–8Na2O–7LaF3 (mol%), which exhibits a lower viscosity and higher kinetic fragility than the studied glass. α-KLaF4 crystallises at 580 °C forming nanocrystals of around 20 nm, which are composed of domains of ~9 nm which vary little in size with time of treatment. The fraction of α-KLaF4 crystals in the glass-ceramic obtained at 580 °C after 150 hours was estimated to be ~5.8 wt%. At higher temperatures, the additional crystallisation of β-KLaF4 takes place (~2.1 wt%.); however, the quantity of α-KLaF4 crystals remains constant. TEM analysis shows that the hexagonal phase forms from the metastable cubic phase and, additionally, smaller α-KLaF4 crystals precipitate from the fluorine remaining in the glassy matrix. The periphery of the precipitated crystals was confirmed by EDX analysis to be enriched in silica, although the additional formation of an alumina shell cannot be excluded. Both shells would constitute a diffusional barrier which limits the crystal growth. The hexagonal phase is formed in conditions of high temperatures, where the influence of this barrier is diminished.The presence of nuclei containing K–F–La bonds in the parent glass, which give rise to α-KLaF4 nano-crystals of cubic symmetry on heat treatment, has been confirmed by 19F MAS NMR. Such nuclei are consistent with a bulk crystallisation mechanism from a constant number of nuclei, as supported by DSC analysis, although a diffusional process from Al–F–Al and F–K environments to the precipitated KLaF4 also takes place.
The presence of two different nanocrystalline structures may be very interesting for optical applications when these materials are doped with rare-earth ions.
Acknowledgements
The authors acknowledge the financial support of FP6 Project INTERCONY, contract no. NMP4-CT-2006-033200, and the CiCyt project MAT2010-20459, as well as the FhG Internal Programs under grant no. Attract 692 280.A. de Pablos-Martín is grateful to the CSIC for a JAE contract 2008–2011.
References
- Q. Luo, X. Qiao, X. Fan, B. Fan and X. Zhang, J. Am. Ceram. Soc., 2011, 94, 1670–1674 CrossRef CAS.
- C. Li, S. Xu, R. Ye, D. Deng, Y. Hua, S. Zhao and S. Zhuang, Phys. B, 2011, 406, 1698–1701 CrossRef CAS PubMed.
- J. Zhang, C. Mi, H. Wu, H. Huang, C. Mao and S. Xu, Anal. Biochem., 2012, 421, 673–679 CrossRef CAS PubMed.
- J.-C. Zhou, Z.-L. Yang, W. Dong, R.-J. Tang, L.-D. Sun and C.-H. Yan, Biomaterials, 2011, 32, 9059–9067 CrossRef CAS PubMed.
- R. E. Thoma, G. M. Hebert, H. Insley and C. F. Weaver, Inorg. Chem., 1963, 2, 1005–1012 CrossRef CAS.
- W. Shao, R. Hua, W. Zhang, Y. Tian, J. Zhao, L. Na, J. Yu and Z. Sun, Powder Technol., 2013, 237, 326–332 CrossRef CAS PubMed.
- J. Guo, F. Ma, S. Gu, Y. Shi and J. Xie, J. Alloys Compd., 2012, 523, 161–166 CrossRef CAS PubMed.
- G. Wang, W. Qin, J. Zhang, L. Wang, G. Wei, P. Zhu and R. Kim, J. Alloys Compd., 2009, 475, 452–455 CrossRef CAS PubMed.
- R. Chai, H. Lian, Z. Cheng, C. Zhang, Z. Hou, Z. Xu and J. Lin, J. Colloid Interface Sci., 2010, 345, 262–268 CrossRef CAS PubMed.
- A. C. Yanes, A. Santana-Alonso, J. Mendez-Ramos, J. del-Castillo and V. D. Rodriguez, J. Alloys Compd., 2009, 480, 706–710 CrossRef CAS PubMed.
- F. Liu, Y. Wang, D. Chen and Y. Yu, Mater. Sci. Eng., B, 2007, 136, 106–110 CrossRef CAS PubMed.
- S. Zhao, S. Xu, G. Jia, D. Deng, L. Huang and H. Wang, Mater. Lett., 2011, 65, 2407–2409 CrossRef CAS PubMed.
- A. Herrmann, M. Tylkowski, C. Bocker and C. Rüssel, Chem. Mater., 2013, 25, 2878–2884 CrossRef CAS.
- M. Sroda, J. Therm. Anal. Calorim., 2009, 97, 239–243 CrossRef CAS PubMed.
- A. de Pablos-Martin, G. C. Mather, F. Muñoz, S. Bhattacharyya, T. Höche, J. R. Jinschek, T. Heil, A. Duran and M. J. Pascual, J. Non-Cryst. Solids, 2010, 356, 3071–3079 CrossRef CAS PubMed.
- A. de Pablos-Martin, N. Hemono, G. C. Mather, S. Bhattacharyya, T. Höche, H. Bornhöft, J. Deubener, F. Muñoz, A. Duran and M. J. Pascual, J. Am. Ceram. Soc., 2011, 94, 2420–2428 CrossRef CAS.
- A. de Pablos-Martin, M. O. Ramirez, A. Duran, L. E. Bausa and M. J. Pascual, Opt. Mater., 2010, 33, 180–185 CrossRef CAS PubMed.
- A. de Pablos-Martin, C. Patzig, T. Höche, A. Duran and M. J. Pascual, CrystEngComm, 2013, 15, 6979–6985 RSC.
- N. Tyagi, A. A. Reddy and R. Nagarajan, Opt. Mater., 2010, 33, 42–47 CrossRef CAS PubMed.
- Y. P. Du, Y. W. Zhang, L. D. Sun and C. H. Yan, Dalton Trans., 2009, 8574–8581 RSC.
- T. G. Filatova, B. S. Zakharova and L. P. Reshetnikova, Vestn. Mosk. Univ., Ser. 2: Khim., 1987, 28, 603–605 CAS.
- W. H. Zachariasen, J. Am. Chem. Soc., 1948, 70, 2147–2151 CrossRef CAS.
- S. Ahmad, G. V. Prakash and R. Nagarajan, Inorg. Chem., 2012, 51, 12748–12754 CrossRef CAS PubMed.
- C. P. Groen, A. Oskam and A. Kovacs, Inorg. Chem., 2003, 42, 851–858 CrossRef CAS PubMed.
- H. Scholze, Ver. Dtsch. Keram. Ges, 1962, 391, 63–68 Search PubMed.
- J. Rodriguez Carvajal, Phys. B, 1993, 192, 55–69 CrossRef CAS.
- T. Höche, J. W. Gerlach and T. Petsch, Ultramicroscopy, 2006, 106, 981–985 CrossRef PubMed.
- S. Bhattacharyya, T. Höche, K. Hahn and P. A. van Aken, J. Non-Cryst. Solids, 2009, 355, 393–396 CrossRef CAS PubMed.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- W. H. G. Tammann, Z. Anorg. Allg. Chem., 1926, 156, 3 Search PubMed.
- H. Vogel, Phys. Z., 1921, 22, 1 Search PubMed.
- I. Avramov and A. Milchev, J. Non-Cryst. Solids, 1988, 104, 253–260 CrossRef CAS.
- J. O. Isard, J. Non-Cryst. Solids, 1969, 1, 235–261 CrossRef CAS.
- C. A. Angell, J. Non-Cryst. Solids, 1991, 131–133, 13–31 CrossRef CAS , Part 1.
- C. Bocker, S. Bhattacharyya, T. Höche and C. Rüssel, Acta Mater., 2009, 57, 5956–5963 CrossRef CAS PubMed.
- W. H. Zachariasen, Acta Crystallogr., 1948, 1, 4 CrossRef.
- W. H. Zachariasen, Acta Crystallogr., 1949, 2, 3 Search PubMed.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louer and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- G. W. Brindley, Phil. Mag. Ser., 1945, 7, 347–369 Search PubMed.
- F. Muñoz, A. de Pablos-Martin, N. Hemono, M. J. Pascual, A. Duran, L. Delevoye and L. Montagne, J. Non-Cryst. Solids, 2011, 357, 1463–1468 CrossRef PubMed.
- T. J. Kiczenski and J. F. Stebbins, J. Non-Cryst. Solids, 2002, 306, 160–168 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- C. Bocker, I. Avramov and C. Rüssel, Scr. Mater., 2010, 62, 814–817 CrossRef CAS PubMed.
- I. Avramov, C. Rüssel and K. Avramova, J. Non-Cryst. Solids, 2004, 337, 220–225 CrossRef CAS PubMed.
- S. Bhattacharyya, T. Höche, J. R. Jinschek, I. Avramov, R. Wurth, M. Müller and C. Rüssel, Cryst. Growth Des., 2010, 10, 379–385 CAS.
- M. J. Pascual, C. Lara and A. Duran, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2006, 47, 572–581 CAS.
- T. Ozawa, J. Therm. Anal., 1976, 9, 369–373 CrossRef CAS.
- T. Ozawa, J. Therm. Anal., 1970, 2, 301–324 CrossRef CAS.
- K. Matusita and S. Sakka, J. Non-Cryst. Solids, 1980, 38–39, 741–746 CrossRef CAS.
- I. W. Donald, J. Non-Cryst. Solids, 2004, 345, 120–126 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2013 |