DOI:
10.1039/C3CE41064A
(Paper)
CrystEngComm, 2013,
15, 9521-9529
Methanol mediated crystal transformations in a solvatochromic metal organic framework constructed from Co(II) and 4-(4-pyridyl) benzoate†
Received
8th June 2013
, Accepted 23rd July 2013
First published on 23rd July 2013
Abstract
The structural transformations of compound 1 {[Co(44pba)2]4·(DMF)3·(EtOH)0.25·(H2O)4}n [44pba = 4-(4-pyridyl) benzoate] were followed during the exchange of DMF and ethanol molecules by methanol at room temperature. The new phases obtained 2 {[Co(44pba)2]·(MeOH)2.5·(H2O)}n, 3 {[Co(44pba)2(MeOH)2]2·(MeOH)2.5·(H2O)2}n and 4 {[Co(44pba)2(MeOH)2]·(MeOH)0.5·(H2O)0.5}n were studied using differential scanning calorimetry, thermogravimetric analysis and X-ray diffraction methods. The final phase 4 reverts back to 1 upon soaking 4 in a dry DMF–ethanol mixture as evidenced by the powder diffraction data and infrared spectra.
Introduction
The study of metal organic frameworks or coordination polymers has attracted the attention of solid state chemists owing to the diverse functionalities and properties that these materials offer, which include sensing, storage, separation and drug delivery.1–6 Besides synthesizing metal organic frameworks or coordination polymers with beautiful topologies and useful properties, one of the goals of scientists working in this area is to understand the mechanisms of the solid state crystal transformation. Such an understanding will make it possible to design reactive solids and conduct reactions in the solid state with a greater ease than before.7 Reactions in the solid state are not unknown as evidenced by the number of organic reactions that have been carried out. The mechanisms that are involved in these reactions are not simple and the majority of them are bimolecular in nature.8,9 Tracking a single crystal to single crystal transformation opens routes for the systematic study of solid state reactions, since it allows a visualization of how the crystal structure changes during the transformation process. These transformations can be very difficult to follow since single crystallinity is not easily retained, on the movement of atoms.10 Recently, a great deal of attention has been focused on the transformation of metal organic frameworks which exhibit different physical properties after single crystal to single crystal transformations.11
Metal organic frameworks have been grouped into different generations depending on the way they behave upon guest removal. Reversible guest induced reformation has been observed in flexible porous frameworks where the removal of the guest molecules induces a structural change in the network, however it reverts to the original structure under the initial conditions. Such compounds have been classified as the third type generation by Kitagawa.12 Structural transformations as a result of guest exchange have been observed in several systems.13–17 Herein, we report a series of structural rearrangements in the framework of a highly porous metal organic framework previously described by our group.18 These rearrangements are triggered by guest exchange and the structural transformation is fully reversible upon the exposure of the final phase to a DMF–ethanol solvent mixture. Previous studies on this compound showed that the material exhibited both solvatochromic and thermochromic properties. In this study we have taken advantage of the X-ray diffraction techniques to follow the structural transformations of the compound that are mediated by methanol.
Experimental
All chemicals were obtained from commercial sources and were used without further purification.
Synthesis of 1: {[Co(44pba)2]4·(DMF)3·(EtOH)0.25·(H2O)4}n (where 44pba is 4-(4-pyridyl) benzoate)
80 mg of 4-(4-pyridyl) benzoic acid (0.4 mmol) was dissolved in 5 ml DMF with heating and stirring. 29 mg of Co(NO3)2·6H2O (0.1 mmol) was dissolved in 2 ml ethanol with stirring. The two solutions were mixed and sealed in a 15 ml Teflon lined autoclave and heated at 105 °C for three days, followed by cooling to room temperature. Purple prismatic crystals were obtained. The analytical data and crystal structure of 1 were previously reported by us.18
Compound 2: {[Co(44pba)2]·(MeOH)2.5·(H2O)}n
Crystals of 2 were obtained by soaking compound 1 in methanol for 24 hours. Elemental analysis % calculated C 57.52, N 5.10, H 5.06. % Found C 57.27, N 5.13, H 4.99.
Compound 3: {[Co(44pba)2(MeOH)2]2·(MeOH)2.5·(H2O)2}n and 4 {[Co(44pba)2(MeOH)2]·(MeOH)0.5·(H2O)0.5}n
These crystals were obtained concomitantly after soaking compound 1 in methanol for four weeks and they were identified based on their colour. 3 was light pink and 4 was dark pink. Elemental analysis for 4: % calculated C 58.47, N 5.00, H 5.15. % Found C 59.81, N 5.13, H 4.03. Insufficient material precluded further analysis for 3.
Thermogravimetric analysis was carried out using a TA Instrument TA-Q500 instrument. In a typical experiment 1–5 mg of the sample was dried on filter paper, placed in open aluminium pans and heated in a dry air atmosphere of nitrogen (50 ml min−1) at a heating rate of 10 °C min−1 within the temperature range of 25–500 °C.
In order to determine the onset temperature of guest loss DSC was performed on all compounds. The samples were dried on filter paper and a sample mass of 0.5–1.5 mg was placed in ventilated aluminium pans with lids and heated at 10 °C min−1. The experiment was performed using a TA Instrument DSC-Q200 under a dry atmosphere of nitrogen gas (50 ml min−1).
Powder X-ray diffraction
Powder X-ray diffraction studies were performed on a HUBER Imaging Plate Guinier Camera 670 with a Ni-filtered Cu Kα radiation (λ = 1.5406 Å) generated by a Philips X-ray Generator at 20 mA and 40 kV. The crystals were applied directly onto Mylar® film and exposed to the radiation for 60 min.
Pawley fitting
X-ray powder data of regenerated 1 (obtained by soaking 4 in DMF–ethanol for 72 h) were recorded using a Bruker D8 advance diffractometer on a zero background sample holder using copper radiation (λ = 1.5406 Å) at 298 K. The crystallographic data for compound 1 were inserted in the phase details section and were allowed to refine. Profile fitting was performed using the PV-TCHZ pseudo-Voigt function included in the peak picking routine in TOPAS.19 The range in 2θ of 0 to 25° was used for profile fitting. The Pawley fit20 method was used to validate the unit cell parameters by profile matching using the ‘hkl-phase’ refinement in TOPAS. The 5th order Chebychev function was used to model the background and a parameter was included to account for sample displacement caused by the zero background sample holder.
Crystal structure determinations
Structure determinations were performed by single crystal X-ray diffraction using a Bruker KAPPA APEX II DUO diffractometer equipped with a graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Data collections were performed at 173 K using a Cryostream cooler (Oxford Cryosystems, UK). The program SAINT21 was used for unit cell refinement and data reduction. Data were corrected for Lorentz-polarization effects and for absorption (program SADABS).22 Structure solutions were achieved by direct methods (program SHELXS)23 and refined by full-matrix least-squares on F2 using SHELXL24 within the X-SEED25 interface. The non-hydrogen atoms were located in the difference electron density maps and were refined anisotropically while all the hydrogen atoms were placed with geometric constraints and refined with isotropic temperature factors. Hydrogen atoms of the solvent molecules could not be located and some of the non-hydrogen atoms of the guest molecules were refined isotropically. Table 1 gives the crystallographic data and refinement parameters. The structures were deposited at the Cambridge Crystallographic Data Centre and allocated numbers: CCDC 931535–931537.
Table 1 Crystallographic parameters of compounds 2–4a
| |
Compound 2 |
Compound 3 |
Compound 4 |
| For compound 1 a = 31.982(1) Å, b = 31.982(1) Å, c = 25.332(1) Å, space group I4. This compound has been published18 and full crystallographic information can be obtained from the Cambridge Structural Database (CCDC 850072). |
| Molecular formula |
CoC26.5H28N2O7.5 |
Co2C54.5H62N4O16.5 |
CoC26.5H27N2O7 |
| Molecular mass |
553.4 |
1096.0 |
544.4 |
| Crystal size/mm3 |
0.22 × 0.21 × 0.20 |
0.45 × 0.27 × 0.20 |
0.14 × 0.13 × 0.08 |
| Temp. of collection/K |
173(2) |
173(2) |
173(2) |
| Crystal symmetry |
Tetragonal |
Monoclinic |
Trigonal |
| Space group |
P4212 |
Pn |
P32 |
| a/Å |
22.659(1) |
17.0218(5) |
10.7022(4) |
| b/Å |
22.659(1) |
10.5990(3) |
10.7022(4) |
| c/Å |
12.704(1) |
18.3064(5) |
22.874(2) |
| β |
90 |
106.073(1) |
90 |
| Z |
8 |
2 |
3 |
| Volume Å3 |
6522.4(8) |
3173.6(2) |
2268.9(2) |
| Dc/g cm−3 |
1.123 |
1.208 |
1.190 |
| 2θ range |
1.27–28.32 |
1.45–28.29 |
2.20–24.77 |
| No. of reflections collected |
43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 343 343 |
57343 |
9657 |
| No. unique reflections |
8130 |
15725 |
2578 |
| No. reflections with I > 2σ |
6087 |
12839 |
1836 |
| Data/parameters refined |
8130/319 |
15725/715 |
2578/320 |
| Goodness of fit, S |
1.051 |
1.074 |
1.298 |
| Final R1 (I > 2σI) |
0.0666 |
0.0569 |
0.1046 |
| Final wR2 (all reflections) |
0.2347 |
0.1951 |
0.3235 |
| Min, max e density/e Å−3 |
−1.21, 1.06 |
−0.68, 1.39 |
−0.92, 0.67 |
Topological studies
The network topology in 1, 2, 3 and 4 was analysed using X-SEED, SYSTRE26 and TOPOS27 and checked against the Reticular Chemistry Structural Resource (RSCR).28 The relevant output files are given in the ESI.†
Results and discussion
Crystal structure description and thermal analysis
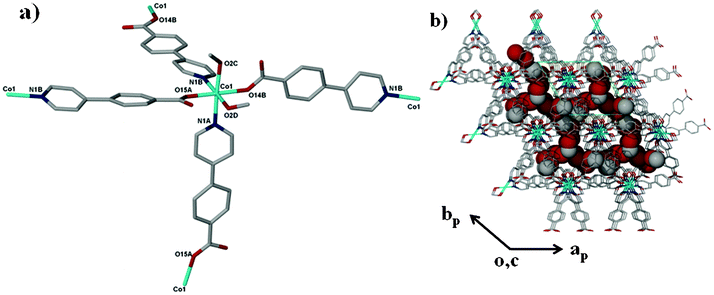
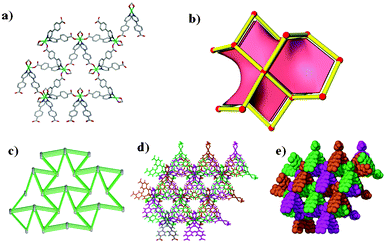
The crystal structure of compound 1 has been described fully in our previous paper.18 For the sake of comparison, its structure is given in the ESI,† Fig. S1. Both 1 and 2 have the same dia topology. The calculated point symbol and vertex symbol for 1 and 2 were found to be 66 and 62.62.62.62.62.62 respectively. Compound 2 crystallizes in the tetragonal crystal system space group P4212. The asymmetric unit of 2 comprises one cobalt centre, two 4-(4-pyridyl) benzoate (44pba) ligands, 2.5 methanol molecules disordered over three positions and a total of 1.1 water molecules disordered over three positions. Each cobalt centre is coordinated to four oxygen atoms and two nitrogen atoms to give rise to a distorted octahedral geometry as illustrated in Fig. 1. The 44pba assumes a μ3-tridentate binding mode to link two cobalt centres. The cobalt to oxygen and cobalt to nitrogen bond lengths are within the reported values in the literature. The relevant bond angles and bond lengths are given in Tables S1 and S2 of the ESI.† Hydrogen bonding interactions exist between the host framework and the water molecules (Ow1⋯O14B = 2.838 Å and Ow2⋯O15A = 3.008 Å). The overall structure has a diamondoid network which is four fold interpenetrated due to the spacious nature of the single network (Fig. 2). Despite this interpenetration, the framework is still highly open, containing channels which run parallel to the c-axis. These channels occupy 46.5% (3029.9 Å3) of the unit cell volume (6522.4 Å3) as estimated in PLATON.29 Thermal analysis by TGA shows a 16.7% initial weight loss which corresponds to the loss of the water and methanol guest molecules from the host channels. For the bulk material having the composition of {[Co(44pba)2]·(MeOH)2.5·(H2O)}n, the solvent content corresponds to a 17.7% mass loss which is a reasonably good match. The decomposition of the guest free compound occurs above 400 °C which suggests that the compound is thermally stable. This can be attributed to the ring catenation that is exhibited in the diamondoid network. As has been noted before, the solvent content can differ from one crystal to another, and for the crystal selected for analysis 2.5 methanol molecules and 1.1 water molecules in total were modelled. In this crystal structure, solvent-accessible voids are still apparent, but no further solvent appears to have been included in this individual crystal. We note however that the unit cell dimensions of 2 correspond closely to those of the recently reported analogous compound {[Co(44pba)2]·(MeOH)4}n.30
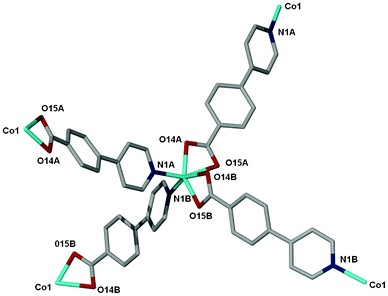 |
| | Fig. 1 The coordination environment of the Co(II) ion in 2. The labelling shows the atoms that are bonded to the cobalt ion. | |
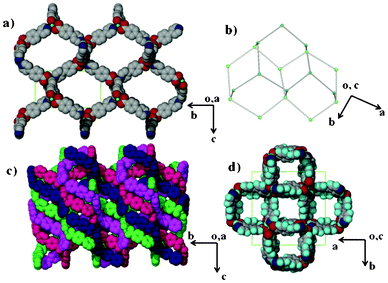 |
| | Fig. 2 (a) A single diamondoid network of 2 viewed along the a-axis. (b) A topological representation of the dia net found in 2. (c) The four fold interpenetration of the network. (d) Notably, even with this interpenetration the framework is highly porous as viewed along the c-axis. The lattice water and methanol molecules occupy these channels and have been omitted for clarity. | |
Compound 3 crystallises in the monoclinic crystal system space group Pn. There are two cobalt centres Co(1) and Co(2) in the asymmetric unit with different coordination environments as shown in Fig. 3. Co(1) is coordinated to four 44pba ligands via two nitrogen atoms of the pyridyl groups, two oxygen atoms (the monodentate binding mode of two carboxylate groups) and two methanol molecules occupying the cis-position. The two pyridyl moieties are coordinated to Co(1) in a cis fashion while the carboxylate groups are trans to each other. Co(2) is coordinated to four 44pba ligands and two methanol molecules but in this case the methanol molecules occupy the trans-position, the pyridyl groups are trans to each other and the carboxylate moieties are bound in the same configuration as in Co(1). The overall coordination geometry around each cobalt centre is distorted octahedral. In addition to the coordinated solvent molecules, a number of uncoordinated solvent molecules were modelled. In the asymmetric unit, we modelled a total of 2.45 uncoordinated methanol molecules disordered over five positions and 1.90 water molecules disordered over four positions. The bond lengths around each cobalt atom are comparable: the Co–N bond length is in the range of 2.147(5)–2.136(6) and the Co–O bond distance is between 2.029(5) and 2.147(5) Å. Detailed information on the bond lengths and bond angles is given in the ESI,† Tables S3 and S4. The total solvent accessible volume in the framework, in the absence of water, methanol and bound guest molecules, is about 44.7% of the unit cell volume as estimated in PLATON. Topologically, 3 is a (4,4)-connected binodal net. Each vertex is 4-connected (Fig. 4). The four connected square planar node which corresponds to Co(2) shown in blue has a point symbol of 42.84 and vertex symbol of 4.4.82.82.88.88, while the tetrahedral node shown in pink which corresponds to Co(1) has the same point symbol of 42.84 but a different vertex symbol of 4.4.87.87.87.87.87. Fig. 4b shows the ideal form of the net of high symmetry and low density drawn with its natural tiling (natural tiles are natural cages in the structure and their faces are the essential rings in the structure). The transitivity of the tiling is 2132. The pts net with large cavities interpenetrates another identical one, giving rise to a 2-fold interpenetrated network of 3 as shown in Fig. 4c and 4d. The interpenetration can be regarded as space filling to stabilize the 3D framework. Insufficient material precluded further studies.
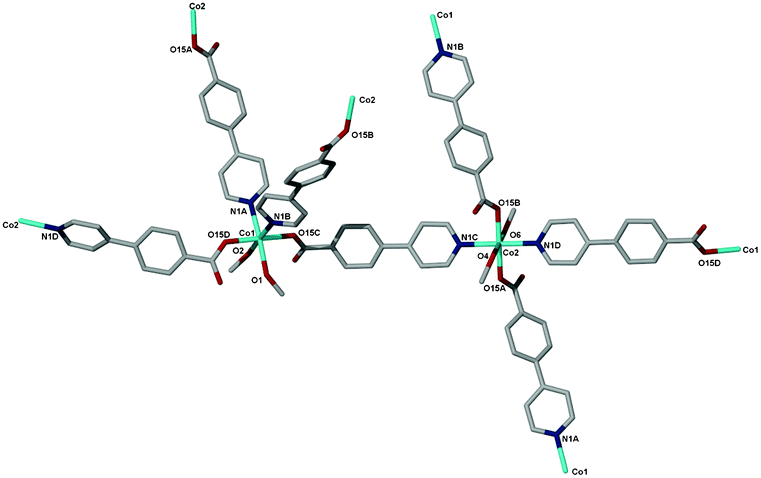 |
| | Fig. 3 The coordination environments of the two cobalt centres Co(1) and Co(2) in 3, the hydrogen atoms, water and methanol guest molecules have been omitted for clarity. | |
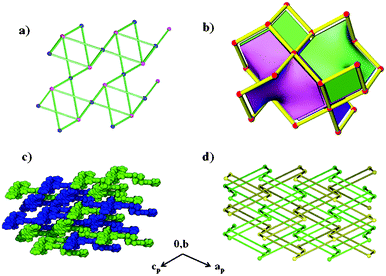 |
| | Fig. 4 (a) A single pts 4,4-connected binodal net found in 3. (b) The ideal form of the net of high symmetry and low density drawn with its natural tilings. (c) The double interpenetrating pts net drawn with van der Waals radii. (d) A topological representation of the double interpenetrating pts net. | |
Compound 4 crystallizes in a trigonal crystal system with the P32 space group. The cobalt(II) centre has a similar geometry as Co(1) in 3 (Fig. 5a). The bond angles and bond lengths around the metal centre are given in Tables S5 and S6 of the ESI.† In the asymmetric unit of 4 a total of 0.4 uncoordinated methanol molecules have been modelled and are disordered over two positions. Two uncoordinated water molecules with a total site occupancy of 0.70 were also modelled in the crystal structure. Extensive hydrogen bonding interactions exist between the water and uncoordinated methanol molecules which reside in the interconnected channels (Fig. 5b). The estimated channel volume in the absence of bound methanol molecules and uncoordinated guest molecules is 37.1% of the unit cell volume. The DSC analysis shows a broad endotherm below 150 °C which corresponds to the loss of water and both bound and unbound methanol molecules as seen in the TGA. The calculated mass loss of 16.36% is in good agreement with the observed loss of 17.46%. The TGA and DSC analysis shows that the compound is stable up to 400 °C. A topological analysis of the framework in 4 revealed a 4-connected uninodal net of qtz (quartz) topology which is three fold interpenetrated. The calculated point symbol and vertex symbol were found to be 64.82 and 6.6.62.87.87 respectively. Fig. 6 shows the qtz net with varying degrees of interpenetration. Interpenetration reduces significantly the size of the triangular channels found in one network to smaller triangular channels in the threefold interpenetrated network as illustrated in Fig. 6d and 6e. Since the channels in 4 are interconnected forming a hexameric network, the triangular channels that are seen along the c-axis may be regarded as windows which may facilitate guest exchange.
 |
| | Fig. 5 (a) The coordination environment of Co(II) in 4. The labelling shows the atoms that are coordinated to the Co(II) ion. (b) The packing diagram of compound 4 as viewed along the c-axis illustrating the presence of the interconnected channels. The channels are occupied by methanol and water molecules drawn with van der Waals radii. | |
 |
| | Fig. 6 The packing and topology in 4: (in all figures, the uncoordinated solvent molecules were omitted for clarity). (a) The single quartz network drawn in the stick form. (b) The ideal form of the quartz net drawn with its natural tiling. The transitivity of the tiling is 1111. (c) A topological representation of the network. (d) The threefold interpenetration of the framework drawn in stick form. (e) The threefold interpenetration drawn using van der Waals radii. | |
Methanol induced phase transformations
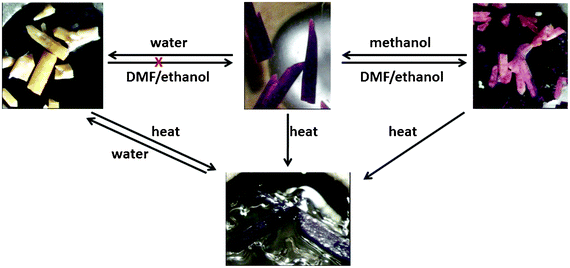
Guest induced structural transformations were studied by thermal analysis and X-ray diffraction methods. Powder X-ray diffraction and single crystal data were collected on a weekly basis to monitor the phase transformations. Fig. 7 shows an illustration of the framework transformations along with photographs of the crystals during this process. Single crystal X-ray diffraction revealed that upon soaking 1 in dry methanol, the guest DMF and ethanol molecules are readily exchanged for methanol molecules as there are no interactions between the guest molecules and the host framework in 1. This occurs via a single crystal to single crystal transformation process. There is a change in the space group from I4 in 1 to P4212 in 2 and a slight rearrangement of the framework from the starting material 1 to compound 2 which does not affect the network topology. Both 1 and 2 have the same connectivity and are made from the same building blocks but they crystallise in different space groups and hence can be regarded as structural supramolecular isomers. The maximum dihedral angle between the pyridyl and the benzene ring in 1 is 40.9° while that in 2 is 33.4° (see Table S7, ESI†). More differences arise in the Co–O bond distances, 1 exhibits the longest distance of 2.373 Å and 2 has a maximum Co–O bond length of 2.302 Å. All these factors give enough evidence that the transformation from 1 to 2 was accompanied by a dynamic rotation of both the pyridyl and the benzene ring, as well as a bond length adjustment allowing for the exchange of guest molecules. Fig. 8 shows the PXRD patterns that were obtained from day one up to day 28. At day 21, a mixture of 2 (purple) and other crystal forms was observed, which could be distinguished by their different colours. At day 28, two crystal forms (3 and 4) were identified based on their colour, 3 was light pink and 4 exhibited a dark pink colour. The PXRD shows that the predominant phase was 4 as it matches well with the simulated PXRD pattern of the single crystal diffraction data. The most significant change from 2 to 3 and 4 are the methanol molecules which become coordinated to the cobalt ion and the carboxylate moiety which changes from a chelating binding mode in 2 to a monodentate mode in both 3 and 4. This suggests that the structural transformation occurs via a cooperative movement of atoms involving the breaking of two metal oxygen bonds and the formation of two new metal–oxygen bonds with the guest molecules. This process resulted in a colour change from purple to pink. The generation of coordinatively unsaturated metal sites by the removal of bound guest molecules has received great attention. Open metal sites promise specific control over the host–guest chemistry which has been recognised as having a potential importance in hydrogen storage.31 Compounds 3 and 4 may find application in this area since they possess coordinated solvent molecules which are readily removed on heating. The powder X-ray diffraction data revealed that the final phase in this transformation process is 4 which was confirmed by PXRD studies after 4 months.
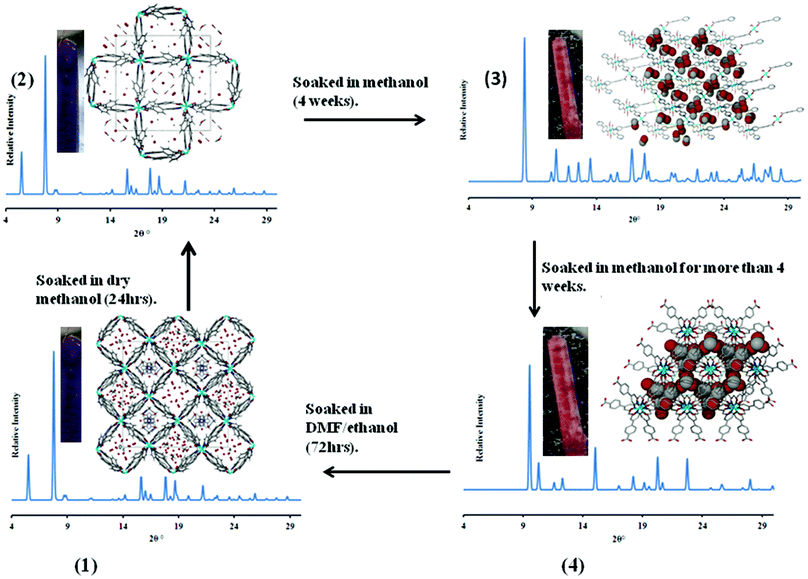 |
| | Fig. 7 An illustration of the structural transformations which compound 1 undergoes. The guest molecules in 1 and 2 are in stick form for DMF and methanol respectively and as red spheres for water molecules. In 3 and 4 the guest molecules are drawn with van der Waals radii. All experiments were carried out at room temperature. | |
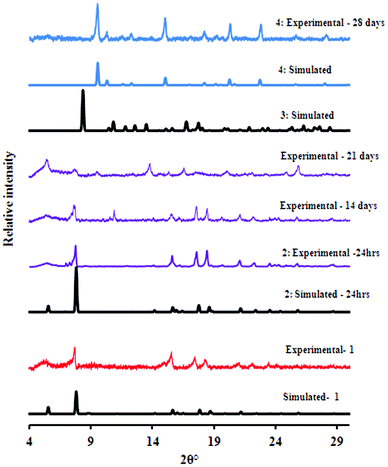 |
| | Fig. 8 The diffraction patterns recorded over a period of 28 days. | |
Although 3 and 4 were obtained at the same time, we believe that 3 is an intermediate phase that quickly transforms into 4. This is due to the fact that 3 has two cobalt centres and one of the centres has the same environment as in 4 (see the preceding section on the structural description). In this regard, we propose that the methanol molecules coordinate first to the cobalt centre in a trans fashion followed by rotation of the methanol molecules around the metal centre to give a cis-conformation in 4. This rotation may be influenced by the nature of the other ligands in the coordination sphere or the metal centre itself. The dynamic motion possible for the monodentate carboxylate with respect to the aromatic ring is illustrated by the change in the torsion angles from 3 to 4 (Table S8, ESI†). In 3 this torsion angle lies in the range of 21–27° while in 4 these moieties are almost co-planar with torsion angles between 1 and 10°. The transformation from 1 to 4 resulted in a general decrease in the Co–O bond length and an increase in the Co–N bond distance (Fig. S3, ESI†).
The framework in compound 1 can be re-established by soaking the final phase 4 in a DMF–ethanol mixture for 72 hours at room temperature. This is confirmed by PXRD and infrared (IR) studies. The IR spectra of compounds 1, 4 and that of 4 after soaking it in a DMF–ethanol mixture are shown in Fig. 9. The IR studies confirm the successful incorporation of DMF during the soaking process as evidenced by the appearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at 1666 cm−1 which is absent in 4 since it does not contain DMF. Similar absorption bands to 1 are also observed after the immersion process which further confirms the reformation of 1. Unfortunately the mosaicity of the crystals increased during this transformation, thus the resultant crystals were not suitable for single crystal data collection. However we confirmed the unit cell of the reconstituted material 1 from 4 (a = 32.081 Å, c = 25.334 Å, space group I4) by whole powder pattern decomposition using the Pawley fitting method in TOPAS (Fig. S4, ESI†).
O band at 1666 cm−1 which is absent in 4 since it does not contain DMF. Similar absorption bands to 1 are also observed after the immersion process which further confirms the reformation of 1. Unfortunately the mosaicity of the crystals increased during this transformation, thus the resultant crystals were not suitable for single crystal data collection. However we confirmed the unit cell of the reconstituted material 1 from 4 (a = 32.081 Å, c = 25.334 Å, space group I4) by whole powder pattern decomposition using the Pawley fitting method in TOPAS (Fig. S4, ESI†).
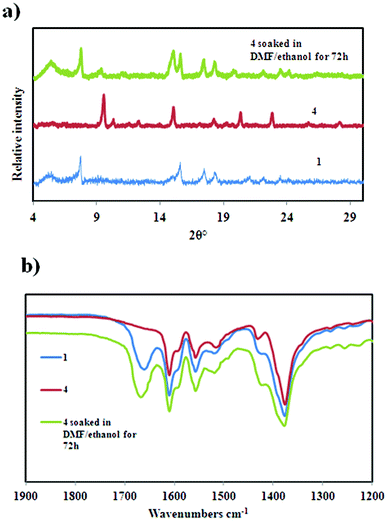 |
| | Fig. 9 (a) The PXRD patterns of 1, 4 and after soaking 4 in a DMF–ethanol solvent mixture for 72 h. (b) The IR spectra confirming the reformation of 1 upon soaking 4 in a DMF–ethanol solvent mixture. | |
Host–guest topology
The topological analysis of the guest molecules that reside in the channels of the host compound is an area that has not yet been explored intensively.32 Some questions to be asked include (i) what insights can such a study provide into the structural rearrangements triggered by the guest molecules? (ii) is there any topological information exchange that exists between the guest molecules and the host compound upon the structural transformation? We believe that the guest topology may have played a role in the reversible structural rearrangement of compound 4 to 1. This prompted us to analyse the host and guest topology independently in compound 4. The guest water and methanol molecules form a hydrogen bonded guest network. We selected a node at one point in this network and the rest of the nodes were then symmetrically generated. Each node is 5-connected and the overall guest connectivity generates a boron nitride topology (bnn). Fig. 10 illustrates the proposed channel–guest templating effect that may have contributed to the reformation of 1. The guest topology in 4 (bnn) and the host topology in 1 (dia) are different. However there exist some similarities in these two types of topologies. One can clearly see that both topologies contain six membered hexagonal rings. The six membered rings in the guest network may have played a role in templating the six membered rings in the host framework of 1. This result is further supported by the reaction cavity concept which states that, among many possible solid state products, the one most likely to be formed is the one which fits best in the cavity that would remain after the reacting molecules are removed from the mother crystal.33 The framework of 1 fits well in the channels of 4 as confirmed by topological analysis.
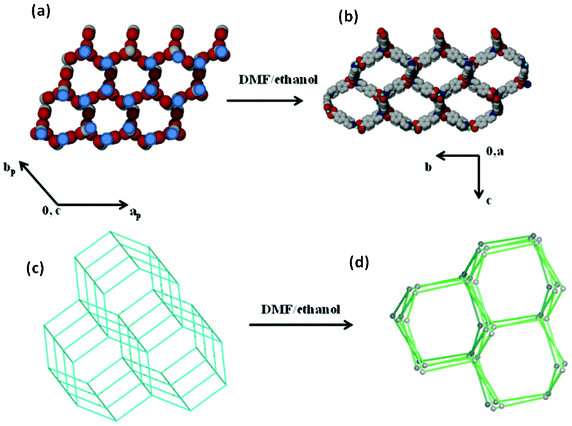 |
| | Fig. 10 A scheme illustrating the channel–guest templating effect that may contribute to the reformation of 1 from 4. (a) The guest connectivity in compound 4; nodes were placed on the blue circles. (b) The framework connectivity in 1. (c) A topological representation of the guest molecules in 4. (d) A topological representation of the host framework in 1. | |
Conclusions
In summary we have observed a series of solvent induced structural transformations in 3D porous frameworks. These structural transformations occurred under very mild conditions (by exposure to methanol and DMF–ethanol at room temperature). The transformations involved the breakage and formation of new bonds around the metal centre with the guest molecules; this resulted in the formation of new structures that could not be afforded by conventional methods. The results obtained in this study coupled with our previous studies18 show that 1 demonstrates interesting properties which could be exploited for chemical sensing and high temperature detection. Fig. 11 summarises these transformations. Potential applications in catalysis and hydrogen storage may be afforded through the post-synthetic modification of 1 to 4. The reformation of 1 from 4 may be attributed to the channel–guest topology. Further studies on host–guest are required to confirm that this has general applicability.
 |
| | Fig. 11 A summary of solvent-mediated structural transformations undergone by compound 1. | |
Acknowledgements
Funding for this project was received from the South African National Research Foundation (NRF), UID 85888. Any conclusions expressed in this article are those of the authors and the NRF does not accept liability in this regard. G. M. is grateful for the financial support he received from the University of Cape Town Chemistry Equity Development Programme and the University of Cape Town 2013 JW Jagger Centenary Gift Scholarship.
Notes and references
- S. R. Batten, S. M. Neville and D. Turner, Coordination Polymers Design, Analysis and Application, Royal Society of Chemistry, Colchester, 2009, vol. 1 Search PubMed.
- D. J. Tranchemontagne, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136 CrossRef CAS.
- G. Ferey, Stud. Surf. Sci. Catal., 2007, 170, 66 CrossRef.
- T. K. Maji and S. Kitagawa, Pure Appl. Chem., 2007, 79, 2155 CrossRef CAS.
- C. Serre, C. Mellot-Draznieks, S. Surble, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS.
- G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal Engineering: A Textbook, World Scientific Pub. Co. Inc., Singapore, 2011 Search PubMed.
- F. Toda, Organic Solid State Reactions, Springer, The Netherlands, 2005 Search PubMed.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS.
-
(a) L. R. MacGillivray, G. S. Papaefstathiou and T. D. Hamilton, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS;
(b) J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781 CrossRef CAS.
-
(a) S. K. Ghosh, J. Zhang and S. Kitagawa, Angew. Chem., Int. Ed., 2007, 46, 7965 CrossRef CAS;
(b) A. Aijaz, E. Barea and P. K. Bharadwaj, Cryst. Growth Des., 2009, 9, 4480 CrossRef CAS;
(c) A. Husain, M. Ellwart, S. A. Bourne, L. Öhrström and C. L. Oliver, Cryst. Growth Des., 2013, 13, 1526 CrossRef CAS.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC.
- M. K. Sharma and P. K. Bharadwaj, Inorg. Chem., 2011, 50, 1889 CrossRef CAS.
- C. Massera, M. Melegari, E. Kalenius, F. Uggozoli and E. Dalacanale, Chem.–Eur. J., 2011, 17, 3064 CrossRef CAS.
- G. J. Halder and C. J. Kepert, Aust. J. Chem., 2006, 59, 597 CrossRef CAS.
- M. P. Suh and Y. E. Cheon, Aust. J. Chem., 2006, 59, 605 CrossRef CAS.
- M. Kawano and M. Fujita, Coord. Chem. Rev., 2007, 251, 2592 CrossRef CAS.
- G. Mehlana, S. A. Bourne and G. Ramon, Dalton Trans., 2012, 41, 4224 RSC.
- A. A. Coelho, TOPAS-Academic, version 4.1 (Computer Software), Coelho Software, Brisbane, 2007 Search PubMed.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357 CrossRef CAS.
- SAINT, version 7.60a, Bruker AXS Inc, Madison, WI, USA, 2006 Search PubMed.
- G. M. Sheldrick, SADABS, version 2.05, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Solution, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- O. D. Friedrichs, Systre 1.1.4beta, http://gavrog.sourceforge.net/, 2007 Search PubMed.
- V. A. Blatov and V. Peaskov, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 457 CrossRef CAS.
-
(a) M. O'Keeffe, M. A. Peskov, M. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef CAS;
(b) M. Ramsden and O. M. Yaghi, Australian National University Supercomputer Facility, Reticular Chemistry Structure Resource, http://rcsr.anu.edu.au/, 2009 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS.
- M.-H. Zeng, Y.-X. Tan, Y.-P. He, Z. Yin, Q. Chen and M. Kurmoo, Inorg. Chem., 2013, 52, 2353 CrossRef CAS.
- S. Ma and L. Meng, Pure Appl. Chem., 2011, 83, 167 CrossRef CAS.
-
(a) S. A. Bourne, J. Lu, B. Moulton and M. J. Zaworotko, Chem. Commun., 2001, 861 Search PubMed;
(b) K. Biradha, A. Mondal, B. Moulton and M. J. Zaworotko, J. Chem. Soc., Dalton Trans., 2000, 21, 3837 RSC.
-
(a) H. E. Zimmerman and E. E. Nesterov, Acc. Chem. Res., 1973, 6, 217–225 CrossRef;
(b) M. D. Cohen, Angew. Chem., Int. Ed. Engl., 1975, 14, 386 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a