 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular 1D ribbons in complexes between a bicyclic-guanidine derivative and di- or monocarboxylic acids†
Vitthal N.
Yadav
and
Carl Henrik
Görbitz
*
Department of Chemistry, University of Oslo, Post box 1033, Blindern, Oslo – 0315, Norway. E-mail: c.h.gorbitz@kjemi.uio.no; Fax: +47 228 55 441
First published on 11th July 2013
Abstract
Zwitterionic crystalline complexes between 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), a guanidine derivative, and two dicarboxylic acids (DCAs) (oxalic acid, adipic acid) as well as a special monocarboxylic acid (glycolic acid) have been analyzed by single crystal X-ray diffraction methods. In the solid state the carboxylic acid forms a monoanion by readily transferring an acidic proton to a TBD base, resulting in formation of strong +N–H⋯O− hydrogen-bonded R22(8) ring motifs, while O–H⋯O interactions expand the network into infinite one-dimensional supramolecular chains. Numerous C(sp3)–H⋯O interactions also contribute in crystal packing, including TBD as a weak donor and O atoms of carboxyl groups or co-crystallized water molecules as acceptors. The hydrogen bonding and crystal packing of all three complexes have been compared with the respective guanidine–carboxylate or related complexes reported previously.
Introduction
The construction of ordered crystalline networks has been the main focus of supramolecular chemistry in recent decades. To achieve this, molecular recognition through intermolecular non-covalent interactions such as H bonds is the foremost crystal engineering approach.1 In this area organic acids and bases are appealing building blocks because of their innate and robust H-bonding complex formation capacity, including N–H⋯O, N⋯H–O and O–H⋯O interactions.2 Unsubstituted guanidine has been extensively incorporated in crystal design and synthesis because of its strong basicity, the unique three-fold structural symmetry of the guanidinium cation, and the ability to form charge-assisted +N–H⋯O− hydrogen bonds with carboxylate, phosphonate, sulphonate and nitro groups etc. that often results in formation of the symmetrical R22(8) H-bonding motif (Fig. 1a)3 in diverse 2D or 3D networks.4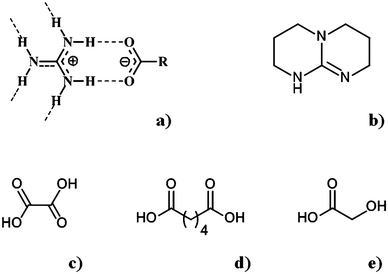 | ||
| Fig. 1 a) A hydrogen bonded guanidinium–carboxylate recognition adduct forming R22(8) motif. b) TBD a base and c) oxalic acid d) adipic acid e) glycolic acid used in complexes. | ||
The guanidine derivative TBD (Fig. 1b) is a rigid and versatile building block that has been widely used as a ligand in co-ordination chemistry5 and often serves as a base6 or a catalyst7 in organic synthesis. Use of TBD in supramolecular chemistry, on the other hand, has been poorly investigated, and only few crystal structures have been reported.3f,7,8 Recently, we have demonstrated that the TBDcation with only two donor sites is in fact a structurally interesting building block when combined with a partner bipyridine dicarboxylate.9 Subsequently we have illustrated different structural properties of TBD–carboxylates via several 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (TBD:DCA) crystalline complexes which form various H-bonded motifs. In these complexes the anti lone pairs of carboxylate O atoms often interact with co-crystallized water molecules by H bonding.10 These waters of hydration lead into well organized water clusters, 1D tapes or open channels rather than the hexagonal networks or densely packed structures obtained in native guanidine complexes.4,9,11
:1 (TBD:DCA) crystalline complexes which form various H-bonded motifs. In these complexes the anti lone pairs of carboxylate O atoms often interact with co-crystallized water molecules by H bonding.10 These waters of hydration lead into well organized water clusters, 1D tapes or open channels rather than the hexagonal networks or densely packed structures obtained in native guanidine complexes.4,9,11
In contrast to the earlier 2:1 complexes, the present work is focusing on unusual solid-state structures formed by TBD (pKa = 14.47)12 with oxalic acid (OxT, Fig. 1c, pKa1 = 1.27; pKa2 = 4.27), adipic acid (AdT, Fig. 1d, pKa1 = 4.41; pKa2 = 5.41) and glycolic acid (GlT, Fig. 1e, pKa = 3.83).13
Experimental
Materials and general process for crystal preparation
TBD, oxalic acid, adipic acid and glycolic acid were purchased from Sigma-Aldrich and used as received. X-ray quality single crystals were obtained by dissolving TBD (2 mmol) and the corresponding carboxylic acid (1 mmol) in demineralized water (2 ml). The mixture was stirred for 10 minutes at ambient temperature and kept stationary to evaporate the solvent slowly over 3–4 weeks. 1H-NMR and 13C-NMR data for the obtained complexes were recorded at RT using a Bruker DPX300MHz spectrometer. Melting point (mp) measurements were performed using Stuart SMP10 equipment.TBD:hydrogen oxalate (OxT)
mp 138–140 °C.1H-NMR, 300 MHz, (DMSO-d6) δ 10.38 (s, 2H), 3.27–3.23 (t, 8H), 3.15–3.11 (t, 8H), 1.90–1.82 (m, 8H).
13C-NMR, 300 MHz, (DMSO-d6) δ, 173.09 (COO), 151.92 (CN3), 46.94 (CH2), 38.06 (CH2), 21.39 (CH2).
TBD:hydrogen adipate (AdT)
mp 238–240 °C.1H-NMR 300 MHz, (DMSO-d6 + D2O) δ, 3.23–3.19 (t, 8H), 3.13–3.09 (t, 8H), 1.94–1.89 (m, 4H), 1.87–1.79 (m, 8H), 1.39–1.34 (m, 4H).
13C-NMR 300 MHz, (DMSO-d6 + D2O) δ, 180.48 (COO), 151.69 (CN3), 46.88 (CH2), 38.60 (CH2), 37.88 (CH2), 26.98 (CH2), 21.11 (CH2).
TBD:glycolate monohydrate (GlT)
mp 88–90 °C.1H-NMR, 300 MHz (DMSO-d6) δ, 9.93 (s, 1H), 3.51 (s, 2H), 3.28–3.24 (t, 4H), 3.17–3.13 (t, 4H), 1.91–1.83 (m, 4H).
13C-NMR 300 MHz (DMSO-d6) δ, 177.29 (COO), 152.04 (CN3), 62.24 (CH2), 46.96 (CH2), 38.05 (CH2), 21.29 (CH2).
X-ray structure determination
X-ray intensity data measurements of OxT, AdT and GlT crystalline complexes were carried out on a Bruker SMART APEX II CCD diffractometer with graphite-monochromatized (Mo Kα = 0.71073 Å) radiation at low temperature (105 K) controlled by an Oxford Cryostream low-temperature device. Data were collected with ω scan width 0.5° at three different settings of φ (0°, 90° and 180°) with the detector fixed at 2θ = −30°. Data integration/reduction and absorption was carried out by SAINT and SADABS, respectively, refinement by SHELXTL.14 In these complexes all the H–C and the H–N hydrogen atoms were constrained to theoretical positions. The carboxylic (O–H) H-atoms of OxT and AdT, and an alcoholic (O–H) and H-atoms of a co-crystallized water molecule in GlT were added from the experimental Fourier difference electron density diffraction map. The GlTwater molecule is observed to be disordered over three sites (O1W, O2W and O3W) with refined occupancies 0.900(7), 0.056(7) and 0.042(4), respectively. The O–H bond lengths of the water molecule were subject to SHELX restraints (see CIF file). The program Mercury15 was used to generate the molecular and packing illustrations of all the complexes. Crystal data are summarized in Table 1.| Crystal | OxT | AdT | GlT | |
|---|---|---|---|---|
| Unit formula | C9H15N3O4 | C13H23N3O4 | C9H17N3O3H2O | |
| Crystal size/mm | 0.90 × 0.30 × 0.10 | 0.40 × 0.20 × 0.15 | 0.55 × 0.30 × 0.10 | |
| Unit weight | 229.23 | 285.37 | 233.25 | |
| Crystal system | Orthorhombic | Monoclinic | Triclinic | |
| Space group | P212121 | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
| a/Å | 7.8795(16) | 7.014(3) | 7.1832(7) | |
| b/Å | 9.760(2) | 12.065(5) | 8.9257(9) | |
| c/Å | 13.612(3) | 17.179(7) | 9.8008(10) | |
| α/° | 90 | 90 | 71.298(1) | |
| β/° | 90 | 90.118(6) | 75.374(1) | |
| γ/° | 90 | 90 | 81.203(1) | |
| V/Å3 | 1046.8(4) | 1453(10) | 574.13(10) | |
| Z | 4 | 4 | 2 | |
| Density/g cm−3 | 1.455 | 1.304 | 1.349 | |
| Absorption coefficient/mm−1 | 0.12 | 0.10 | 0.11 | |
| θ range for data collection/° | 2.6–28.9 | 2.1–28.5 | 2.3–28.7 | |
| Reflections | ||||
| Collected | 9443 | 10766 |
5152 | |
| Independent | 1592 | 3687 | 2989 | |
| Observed | 1536 | 2527 | 2422 | |
| R int | 0.019 | 0.061 | 0.011 | |
| wR(F2) | 0.0710 | 0.133 | 0.094 | |
| R[F2 > 2σ(F2)] | 0.0278 | 0.053 | 0.034 | |
Results and discussion
Complex OxT
The anhydrous orthorhombic crystals of OxT contain a hydrogen oxalate anion and a TBDcation in the asymmetric unit (Fig. 2a). The O1 and O2 atoms of the oxalate are H-bonded to TBD so as to form a symmetrical R22(8) ring motif as shown in Fig. 1a. The carboxyl H-atom (H4O) of the hydrogen oxalate is donated to an anti lone pair of O1 (for details of H-bond data see Table 2). This strong carboxyl–carboxylate (COOH⋯−OOC) interaction leads to formation of an infinite 1D chain of hydrogen oxalate along the a-axis that is decorated by TBDcations (Fig. 2b). The resulting ribbons are densely stacked in a herringbone-like array as shown in Fig. 2c, where the polar hydrogen oxalate chain at the core appears to be encapsulated by the neighboring hydrophobic TBD moieties. | ||
| Fig. 2 a) The asymmetric unit of OxT with atomic label scheme. Displacement ellipsoids are shown at the 50% probability level. b) Capped stick representation of one-dimensional hydrogen bonded molecular ribbon in OxT viewed down the crystallographic c-axis. Hydrogen atoms not involved in hydrogen bonds have been omitted for clarity. c) Three-dimensional herringbone pattern of 1D ribbons in the crystal packing of OxT viewed along the a-axis. At the core, the highlighted light-blue shade represents the encapsulation of polar hydrogen–oxalate chain by hydrophobic TBD molecules. | ||
| D–H⋯A | D–H | H⋯A | D⋯A | ∠(D–H⋯A) |
|---|---|---|---|---|
| a Symmetry codes i) x − 1/2, 1/2 − y, 1 − z; ii) −x + 1, −y + 1, −z + 1; iii) x, −y + 3/2, z + 1/2; iv) −x, 1 − y, 2 − z; v) −x, −y, 2 − z. | ||||
| Crystal OxT | ||||
| N1–H1⋯O1 | 0.86 | 1.98 | 2.839(14) | 179 |
| N3–H3⋯O2 | 0.86 | 1.94 | 2.817(13) | 174 |
| O4–H4O⋯O1i | 0.90(2) | 1.65(2) | 2.549(13) | 172(18) |
| Crystal AdT | ||||
| N1–H1⋯O1ii | 0.88 | 1.92 | 2.796(2) | 179 |
| N3–H3⋯O2ii | 0.88 | 1.85 | 2.723(2) | 173 |
| O4–H4⋯O2iii | 0.99(3) | 1.56(3) | 2.536(2) | 170(3) |
| Crystal GlT | ||||
| N1–H1⋯O1 | 0.88 | 1.98 | 2.855(11) | 176 |
| N3–H3⋯O2 | 0.88 | 1.89 | 2.772(11) | 176 |
| O3–H3O⋯O1iv | 0.85(17) | 1.96(17) | 2.741(11) | 152(14) |
| O1W–H11W⋯O2 | 0.87(2) | 1.93(2) | 2.802(12) | 176(19) |
| O1W–H12W⋯O3v | 0.83(2) | 2.06(2) | 2.887(14) | 174(2) |
Unlike the three-dimensionally H-bonded structure of regular guanidinium–hydrogen oxalate hydrate4a (refcode GUHOXM, Cambridge Structure Database,16CSD v5.34, Nov. 2012), the hydrogen oxalate in OxT is non-planar and exists in a twisted conformation [torsion angle O1–C8–C9–O3 = −54.13(15)°] to facilitate formation of the 1D chain. Such an adapted orientation of hydrogen oxalate in OxT clearly deviates from the theoretical 90° torsion angle of oxalate.17 Thus, the crystal structure of OxT demonstrates that by reducing the H-bond complexity of the guanidinium ion, the molecular assembly can be directed towards a less complicated 1D supramolecular structure.
Complex AdT
The asymmetric unit of AdT (Fig. 3a) and the formation of one-dimensional ribbons driven by carboxyl–carboxylate H bonding (Fig. 3b) are closely reminiscent of the observations made for OxT (Fig. 2a and 2b). However, there are two obvious differences between these two structures: first the carboxylate groups of adipic acid are not directly connected as for oxalic acid, but separated by the –(CH2)4– linker, and second the 1D ribbons of AdT are planar and aligned two-dimensionally in the crystallographic bc-plane to form an interlocked sheet-like topology (Fig. 3b), whereas OxT shows a non-planar herringbone-like pattern. The sheets in AdT are stacked within the π⋯π interplanar distance range ∼3.1–3.7 Å [for (N1–N2–N3)TBD⋯TBD′(N1–N2–N3) and (COO−⋯COO−)] and within the van der Waals space of 2.287–2.394 Å for adipate/TBDC(sp3)–H⋯H–C(sp3)-adipate (Fig. S1, ESI†). | ||
| Fig. 3 a) The asymmetric unit of crystal AdT with atom label scheme. Displacement ellipsoids are shown at the 50% probability level. b) The 1D infinite chains in AdT running along the crystallographic c-axis. These chains are interlocked in a zigzag pattern which forms a layer parallel to plane-bc. The non-hydrogen bonded hydrogen atoms have been omitted for clarity. | ||
The crystallization experiment between the adipic acid and TBD was set up to obtain a 2:1 crystalline complex containing two TBDcations and a dianion of adipic acid. In fact, the resulting AdT crystal contains a monoanion rather than a dianion. To facilitate a second deprotonation step by breaking the strong carboxyl–carboxylate interaction, three additional crystallization experiments with increased TBD concentrations (adipic acid:TBD ratios 1:3, 1:4 and 1:5) and one controlled experiment with 1:1 ratio were carried out. The 1:1 mixture instantaneously afforded the familiar AdT type of crystal, whilst other mixtures did not yield crystals. This apparently confirms that the 1:1 ratio of adipic acid and TBD is the thermodynamically favorable composition for crystal formation.
In theory, a dicarboxylic acid should form a dianion upon proton exchange with a base, though in reality the structural characteristics of the acid and base involved may also promote formation of monoanions. This is clear from a survey of dicarboxylic acids in the CSD, where mono- and dianions of oxalic acid are equally common (∼190 structures each). The distribution between mono- and dianion for succinic acid with a –(CH2)2– linker (Succ− = 48; Succ2− = 65) and adipic acid (Adp− = 13; Adp2− = 34), reveals a trend: when the aliphatic link between the two carboxylic groups is extended, the ratio of mono/dianion decreases.
Complex GlT
The asymmetric unit of the GlT complex is composed of the glycolate anion, TBD and a water molecule (Fig. 4a). The TBD and glycolate ions are H bonded by two +N–H⋯O− interactions forming a R22(8) motif. An TBD–glycolate complex is interacting with another TBD–glycolate through glycolateO–H⋯Ocarboxylate interactions and generating a tetramer through the new R22(10) ring motif, (Fig. 4b). Co-crystallized water molecules serve as links between tetramers through formation of a third hydrogen bonded ring motif, R44(14), thus producing a supramolecular ribbon along the b-axis (Fig. 4b). The compact and staggered spatial arrangement of ribbons in AdT is significantly influenced by the weak interactions such as N1⋯H62–C6 (2.67 Å) and C–H⋯O (see Table 3). | ||
| Fig. 4 a) The asymmetric unit of GlT. Displacement ellipsoids are shown at the 50% probability level. b) One-dimensional infinite molecular ribbon in GlT. Three hydrogen bonded ring motifs, 1, R22(8); 2, R22(10) and 3, R44(14) have been highlighted by light-blue shades. A small circle represents the inversion centre of symmetry. c) The crystal packing of one-dimensional ribbons viewed along the b-axis. Light-blue shade represents the hydrophobic surroundings of adipate:water chain. Hydrogen atoms except H bonded have been omitted for clarity in b) and c). | ||
| Complex | C–H⋯O | C–H⋯O | C⋯O | ∠(C–H⋯O) |
|---|---|---|---|---|
| OxT | C3–H31⋯O4 | 2.54 | 3.258(2) | 128.5 |
| C6–H61⋯O3 | 2.61 | 3.590(2) | 167.0 | |
| AdT | C2–H22⋯O3 | 2.54 | 3.485(4) | 158.9 |
| C6–H62⋯O4 | 2.59 | 3.394(4) | 137.8 | |
| GlT | C5–H51⋯O1 | 2.45 | 3.237(2) | 135.3 |
There is no crystal structure involving native guanidine and glycolic acid in the CSD, but a close structural analogue, the guanidinium–bicarbonate, is reported (refcode: DUMPUW).18 This bicarbonate is a methylene group shorter than the glycolate, but it forms a very similar H-bonded tetramer. As the bicarbonate salt is devoid of water molecules, the tetramers are, however, not linked into chains but generate a compact 3D network (see ESI,† Fig. S2).
C–H⋯O interactions and crystal packing
Although the C–H⋯O hydrogen bond is a weak non-covalent interaction, in molecular self-assembly its role is proficient enough to direct the 3D aggregations. This interaction is formed between C and O atoms within a distance range of 3.0–4.0 Å.19 In the present study the N–H⋯O and O–H⋯O hydrogen bonds are electrostatic and directive in binary crystalline complex formation. The role of the aliphatic skeleton of TBD and its contribution to the crystal packing is more variable due to the conformational flexibility of the fused ring system and the multiple and nondirective weak C–H donor implications. In the OxT, AdT and GlT structures there is a variety of C–H⋯O interactions between the TBDmethylene H atoms and strong H-bond accepting O atoms in either the carboxyl groups (see Fig. 5a and 5b) or water (Fig. 5c), which are greatly involved in creating a densely packed crystalline network. The carboxyl group in OxT coordinates with two TBD from different layers and forms H–O⋯H–C3 and >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–C6 contacts, (bond lengths and angles are summarized in Table 3), where both the TBD orient perpendicularly to the carboxyl plane to avoid steric conflict (Fig. 5a). By contrast, in AdT the carboxyl group of the planar and layered hydrogen adipate is aligned parallel to the plane of TBD, interacting with H–C2 and H–C6 (Fig. 5b). Exceptionally the C–H⋯O interaction in GlT involves an additional solvated water molecule as acceptor rather than a carboxylate which is fully engaged in strong H bonding with the TBD and water (Fig. 5c).
O⋯H–C6 contacts, (bond lengths and angles are summarized in Table 3), where both the TBD orient perpendicularly to the carboxyl plane to avoid steric conflict (Fig. 5a). By contrast, in AdT the carboxyl group of the planar and layered hydrogen adipate is aligned parallel to the plane of TBD, interacting with H–C2 and H–C6 (Fig. 5b). Exceptionally the C–H⋯O interaction in GlT involves an additional solvated water molecule as acceptor rather than a carboxylate which is fully engaged in strong H bonding with the TBD and water (Fig. 5c).
 | ||
| Fig. 5 C–H⋯O interactions formed by TBD with a) carboxyl in OxT, b) and AdT, and c) with water in GlT. | ||
Conclusion
Here we have pursued a crystalline complex formation approach based on the R22(8) synthon formed by dicarboxylic acids and a TBD base. This strategy implies controlling the capacity of a potential multiple H-atom donor such as native guanidinium cation. This restrictive path can generate ordered and relatively less complicated lower order crystalline molecular networks (e.g.OxT and GlT). Complexes building an infinite H-bonded network of 1D-chains or tapes, are of interest for materials such as gelators.20 Hence this method may provide a model for further materials preparation. TBD and its weak aliphatic C–H co-ordination should offer insight into the structurally and biologically relevant interactions of similar groups of molecules.Notes and references
- (a) G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed; (b) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS; (c) C. B. Aakeroy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- (a) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS; (b) O. Félix, M. W. Hosseini, A. De Cian and J. Fischer, Angew. Chem., Int. Ed. Engl., 1997, 36, 102–104 CrossRef CAS; (c) A. Mukherjee and G. R. Desiraju, Chem. Commun., 2011, 47, 4090–4092 RSC; (d) M. Du, Z.-H. Zhang and X.-J. Zhao, Cryst. Growth Des., 2005, 5, 1199–1208 CrossRef CAS; (e) H. P. Jones, R. J. Davey and B. G. Cox, J. Phys. Chem. B, 2005, 109, 5273–5278 CrossRef CAS; (f) J. F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee, H. R. Guzmán and Ö. Almarsson, J. Am. Chem. Soc., 2003, 125, 8456–8457 CrossRef CAS; (g) S.-p. Chen, L.-l. Pan, Y.-x. Yuan, X.-x. Shi and L.-j. Yuan, Cryst. Growth Des., 2009, 9, 2668–2673 CrossRef CAS; (h) D. R. Trivedi and P. Dastidar, Cryst. Growth Des., 2006, 6, 1022–1026 CrossRef CAS; (i) K. Kinbara, Y. Hashimoto, M. Sukegawa, H. Nohira and K. Saigo, J. Am. Chem. Soc., 1996, 118, 3441–3449 CrossRef CAS; (j) J. C. MacDonald, P. C. Dorrestein and M. M. Pilley, Cryst. Growth Des., 2001, 1, 29–38 CrossRef CAS.
- (a) M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS; (b) E. D. Raczyńska, M. K. Cyrański, M. Gutowski, J. Rak, J.-F. Gal, P.-C. Maria, M. Darowska and K. Duczmal, J. Phys. Org. Chem., 2003, 16, 91–106 CAS; (c) J. Han, C.-W. Yau, C.-K. Lam and T. C. W. Mak, J. Am. Chem. Soc., 2008, 130, 10315–10326 CrossRef; (d) W.-Z. Xu, J. Sun, Z.-T. Huang and Q.-Y. Zheng, Chem. Commun., 2009, 171–173 RSC; (e) A. C. Soegiarto, A. Comotti and M. D. Ward, J. Am. Chem. Soc., 2010, 132, 14603–14616 CrossRef CAS; (f) K. Latham, J. E. Downs, C. J. Rix and J. M. White, J. Mol. Struct., 2011, 987, 74–85 Search PubMed.
- (a) J. Adams, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1218–1220 CrossRef; (b) V. Videnova-Adrabinska, E. Obara and T. Lis, New J. Chem., 2007, 31, 287–295 RSC; (c) G. Smith, U. D. Wermuth, D. J. Young and P. C. Healy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o556–o557 Search PubMed; (d) G. Smith and U. D. Wermuth, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, o575–o580 Search PubMed.
- (a) S. H. Oakley, D. B. Soria, M. P. Coles and P. B. Hitchcock, Dalton Trans., 2004, 537–546 RSC; (b) U. Wild, P. Roquette, E. Kaifer, J. Mautz, O. Hübner, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2008, 2008, 1248–1257 Search PubMed; (c) M. P. Coles, Chem. Commun., 2009, 3659–3676 RSC.
- (a) D. Simoni, M. Rossi, R. Rondanin, A. Mazzali, R. Baruchello, C. Malagutti, M. Roberti and F. P. Invidiata, Org. Lett., 2000, 2, 3765–3768 CrossRef CAS; (b) D. J. Phillips and A. E. Graham, Synlett, 2010, 769, 773 Search PubMed; (c) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS.
- C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef.
- (a) I. Binkowska, A. Jarczewski, A. Katrusiak, G. Wojciechowski and B. Brzezinski, J. Mol. Struct., 2001, 597, 101–107 CrossRef CAS; (b) A. Huczyński, M. Ratajczak-Sitarz, A. Katrusiak and B. Brzezinski, J. Mol. Struct., 2008, 888, 84–91 CrossRef CAS.
- V. N. Yadav and C. H. Görbitz, CrystEngComm, 2013, 15, 439–442 RSC.
- V. N. Yadav and C. H. Görbitz, Cryst. Growth Des., 2013, 13, 2174–2180 Search PubMed.
- T. C. W. Mak and F. Xue, J. Am. Chem. Soc., 2000, 122, 9860–9861 CrossRef CAS.
- pKa, Calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (©1994–2013 ACD/Labs).
- H. C. Brown, D. H. McDaniel and O. Häfdinger, Determination of Organic Structures by Physical Methods, ed. E. A. Braude and F. C. Nachod, Academic Press, New York, 1955 Search PubMed.
- (a) Bruker APEX2, SAINT-Plus and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed; (b) G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112–122 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- D. Y. Naumov, E. V. Boldyreva, N. V. Podberezskaya and J. A. K. Howard, Solid State Ionics, 1997, 101–103, 1315–1320 CrossRef CAS.
- D. A. Baldwin, L. Denner, T. J. Egan and A. J. Markwell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1197–1199 CrossRef.
- (a) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond In Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed; (b) G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS; (c) T. Steiner, Crystallogr. Rev., 2003, 9, 177–228 CrossRef CAS.
- R. Luboradzki, O. Gronwald, M. Ikeda, S. Shinkai and D. N. Reinhoudt, Tetrahedron, 2000, 56, 9595–9599 CrossRef CAS.
Footnote |
| † CCDC 905593–905595. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce40960k |
| This journal is © The Royal Society of Chemistry 2013 |