 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The molecular self-association of carboxylic acids in solution: testing the validity of the link hypothesis using a quantum mechanical continuum solvation approach†
Devis
Di Tommaso
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1 H0AJ, United Kingdom. E-mail: d.tommaso@ucl.ac.uk
First published on 19th June 2013
Abstract
According to the “link hypothesis” there is a connection between the solvent-dependent most stable self-associated groups of molecules and the building unit present in the polymorph that crystallizes from solution. We have tested this hypothesis by computing the Gibbs free energies for the molecular self-association in solution of three selected monocarboxylic acids that crystallize in different polymorphic forms and exhibit selective crystallization depending on the solvent: tetrolic acid (TTA), m-aminobenzoic acid (mABA) and m-hydroxybenzoic acid (mHBA). Calculations have been conducted at the density functional theory (M06-2X) level with the SMD polarizable continuum model to simulate aqueous and organic solutions. For all three systems we have found that the solvation environment significantly affects the stability of dimers. The most stable dimer in solution is the classic carboxylic acid dimer, and its stability decreases on going from nonpolar solvents, such as chloroform or acetonitrile, to aqueous and alcoholic solutions. However, whilst for TTA and mABA there is a link between the carboxylic dimer synthon in solution and the structural synthon packed in the metastable α-TTA polymorph, which crystallizes from chloroform, and in the crystal form-II of mABA, which crystallizes from acetonitrile, for mHBA the stabilization of the centrosymmetric carboxylic dimeric in acetonitrile and ethyl acetate does not correspond with recent experimental observations that the polymorph containing this structural unit (form I) is not the crystal form that nucleates preferentially from these solvents (Cryst. Growth Des., 2013, 13, 1140). Starting from the carboxylic dimer of TTA, the processes of trimerization and tetramerization of TTA from solution have been also modelled. The calculations suggest that the formation of tetramers in chloroform occurs from the self-association of the carboxylic acid dimers and not through the association of the TTA monomers and trimers. The free energy formation of the ionized forms of the mABA dimer (non-zwitterionic–zwitterionic and zwitterionic–zwitterionic) has been evaluated and, according to our results, zwitterionic–zwitterionic mABA dimers might be abundant in supersaturated aqueous and alcoholic solutions of m-aminobenzoic acid.
Introduction
Crystallization of molecular crystals is a key industrial process that is closely linked to the phenomenon of polymorphism, that is, the ability of a molecule to crystallise in more than one structure. The pharmaceutical industry in particular requires the production of the desired polymorph as drugs receive regulatory approval for only a single polymorph. Approximately one-third of all active pharmaceutical ingredients are confirmed polymorphic systems,1 and a similar number has been reported for organic compounds in general.2 It is therefore essential to have reliable and effective methods for the selection of each specific polymorphic form of a molecular crystal. However, the lack of fundamental understanding of the factors dictating the outcome of crystallization processes is such that achieving control of a specific polymorph remains a significant challenge.While a variety of mechanisms are thought to play a role, including growth in confinement, adjustment of the precipitating medium and the formation of amorphous precursor phases, it is the solvent that mostly influences the thermodynamics and kinetics of crystal growth processes.3 In fact, the approach that is usually followed to achieve polymorph selection during solution crystallization is the modification of the solvation environment by changing the nature of the solvent or through the addition of additives (organics, peptides or simple ionic salts).4,5 However, this approach for polymorph selection still remains ad hoc and is heavily dependent on trial and error.
At the present there is no general model explaining how the modification of the chemistry of a solution causes the formation of different polymorphs and simple kinetic models such as Classical Nucleation Theory are unable to predict, or even qualitatively explain, the phenomenon of polymorph selection during solution crystallization.6 There are, however, some hypotheses regarding which processes in solution could be responsible for the selective crystallization of organics.
According to the “link hypothesis” solute molecules tend to self-assemble into different structural motifs in solution, known as growth synthons,7 and there is a direct link between the solvent-dependent most abundant pre-nucleation aggregates and the polymorph that crystallizes from solution.8–10 Therefore, if the “link hypothesis” is correct, then from the knowledge of the growth synthons present in solution, as a consequence of thermodynamically driven self-association equilibrium, it should be possible to determine the structural synthons packed in the crystal, as identified by crystallography, and consequently predict the formation of a specific polymorph. For example, if the association to a specific dimer or trimer occurs in solution, then the same building unit should be observed in the crystal.11
Unbiased molecular dynamics (MD) has been the computational method of choice to determine the population statistics of molecular clusters in solution.9,12–16 However, to evaluate the thermodynamic stability of molecular clusters in solution, what is required is to calculate the free energy change associated with the process of molecular association. In molecular dynamics simulations the free energies of association can be computed using umbrella sampling or metadynamics, but these methods require the identification of one or a set of collective variables to span the “reactive” subspace of the chemical system under investigation. With the exception of simple molecular systems, such as tetrolic acid,17 the choice of collective variables associated with the molecular self-assembly process of organic molecules is not always obvious. Moreover, MD simulations of organic molecules in explicit solvent molecules usually rely on classical molecular mechanics force fields, which are unable to account for electronic polarization or proton transfer. For example, contradictory results regarding the stability and structure of the molecular associates of glycine9,12 and urea18,19 in aqueous solutions were obtained using different force fields, and only thanks to the application of ab initio molecular dynamics it was possible to identify the most stable dimers of glycine20 and urea.21 However, this technique is limited by the size and complexity of the system, as well as by the time frame of the simulation.
An alternative first principles method that can be used to compute the free energy of molecular association in solution is based on the quantum mechanical continuum solvation approach,22 in which the solute (monomer, dimer, trimers, etc.) is described at a quantum mechanical level (ab initio or density functional) and the solvent is treated as a polarizable continuum. In particular, the quantum mechanical treatment of molecular associates using modern density functionals specifically developed for non-bonding interactions23,24 gives an accurate description of the hydrogen (H) bonding and van der Waals forces that can be important in polymorph selection at an accessible computational cost. On the other hand, polarizable continuum models represent a practical approach to simulate the solvation environment and determine the effect of the medium on the structure and stability of solutes in solution.
Here, we report the application of this quantum mechanical continuum solvation approach to compute the free energy of association in solution of three monocarboxylic acids that crystallize in different polymorphic forms and exhibit a selective crystallization depending on the solvent: tetrolic acid (TTA), m-aminobenzoic acid (mABA) and m-hydroxybenzoic acid (mHBA) (see Table 1). The objectives of this paper are to assess the effect of the solvation environment on the stability of pre-nucleation molecular aggregates of tetrolic acid, m-aminobenzoic acid and m-hydroxybenzoic acid, and to verify if there is a connection, as suggested by the link hypothesis, between the structures of the most thermodynamically stable molecular self-associate in a certain solvent and the structural synthon of the crystal polymorph that crystallizes from that particular solvent.8
| Polymorphs | Effect or solvent | ||
|---|---|---|---|
| a Ref. 4. b Ref. 37. c Ref. 42. | |||
| Tetrolic acid (TTA) |
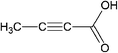
|
α-TTA | α-TTA : CCl4 and CHCl3a |
| β-TTA | β-TTA : alcoholic solutionsa | ||
| m-Aminobenzoic acid (mABA) |
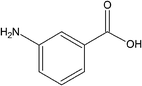
|
I-mABA | I-mABA: water, methanolb |
| II-mABA | II-mABA: acetonitrileb | ||
| III-mABA | |||
| IV-mABA | |||
| V-mABA | |||
| m-Hydroxybenzoic acid (mABA) |
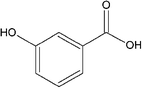
|
I-mHBA | I-mHBA: ethanolc |
| II-mHBA | II-mHBA: ethyl acetatec | ||
Computational methodology
Free energies of association in solution
In the gas phase, the free energy of molecular association has been computed as:| ΔGoass = GoAB − GoA − GoB | (1) |
| GoX = Ee,gas + δGoVRT,gas | (2) |
In the liquid-phase the molecular association has been modelled using the thermodynamic cycle reported in Scheme 1, and the free energy change for the association reaction in solution in the 1 mol L−1 standard state has been computed using the following equation:
| ΔG*ass = G*AB − G*A − G*AB | (3) |
| G*X = GoX + ΔG*solv + ΔG°→* | (4) |
| G*X = ETotsoln + δG*VRT,soln | (5) |
| ETotsoln = Ee,soln + ΔGEP + GCDS | (6) |
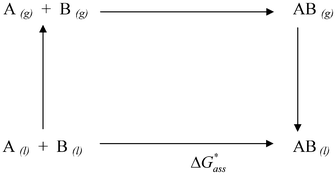 | ||
| Scheme 1 | ||
In the SMD continuum solvation model, which has been used in this study, the GCDS contribution to the free energy has been parameterized in order to take into account solvent effects specific to the first solvation shell.28 Therefore, it should be capable of describing the extra-stabilization in solution associated with the ability of the solute to form hydrogen bonds with the molecules of the solvent. If the calculations are conducted with the code Gaussian09 and the SMD polarizable continuum model, the term ETotsoln corresponds to the basic energy of a density functional theory calculation using the SMD model. The vibrational, rotational and translational contribution to the solution free energy (δG*VRT,soln in eqn (5)) is computed by applying the ideal gas partition functions to the frequencies calculated in the dielectric continuum and the 1 M standard state. This approach has been questioned by Ho et al.26 because ideal gas partition functions are unlikely to be valid in solution, but Ribeiro et al. have demonstrated that vibrational contributions to a solute's free energy are in general insensitive to whether the solute vibrational free energy are computed in the gas-phase or in solution.27
Electronic structure calculations
All calculations were carried out with NWChem 6.129 and Gaussian0930 codes. If the molecular association in solution was modelled using the thermodynamic cycle in Scheme 1, and the related eqn (3) and (4), then the following computational protocol was employed to compute the free energies of association reaction:Gas-phase calculations were performed with the M06-2X hybrid-meta density functional theory (DFT) functional.23 We have chosen M06-2X because its assessment against representative databases showed that this method is one of the most accurate density functionals for a combination of main-group thermochemistry, kinetics and noncovalent interaction.23,31
Optimised structures and frequencies were computed using the 6-31++G(d,p) basis set with the temperature and pressure fixed at the values of 298 K and 1 atm, respectively, and gas-phase vibrational, rotational and translational contributions to the free energy (δGoVRT,gas) were evaluated on the basis of these frequencies and the ideal-gas partition functions q(p,T).
For the evaluation of the term Ee,gas in eqn (2), more accurate energetics were derived from single-point energy calculations with the aug-cc-pVDZ basis set because for the M06-2X method this functional/basis set combination gives the best compromise between accurate binding energies and computational cost.32
The free energies of solvation of the species involved in the association process (ΔG*solv) were calculated using the SMD solvation model,28 along with the M06-2X functional and the aug-cc-pVDZ basis set [SMD/M06-2X/aug-cc-pVDZ] and the optimized gas-phase geometries. SMD is the recommended continuum solvation model by the Gaussian09 manual for computing the free energy of solvation. In fact, the SMD model was originally tested over a set of 2892 solvation free energies and transfer free energies for neutral solutes and ions in water and non-aqueous solutions, and the mean unsigned error (MUE) over 26 combinations of various basis sets and density functionals was 0.8 kcal mol−1 for neutral solutes, and 4.3 kcal mol−1 for ions.33 In particular, the MUE in calculated aqueous solvation free energies of carboxylic acids was only 0.25 kcal mol−1 using the M06-2X/6-31+G(d,p) level of theory. The SMD model together with the M06-2X density functional was also applied to predict the free energies of aqueous solvation for 61 drug-like molecules in the SAMPL1 test set, and the authors reported a MUE of 2.0 kcal mol−1.34 The SMD/M06-2X level of theory was also used by Ribeiro and co-workers to compute the partition coefficients of nucleobases between chloroform and water with a MUE of 0.8 kcal mol−1.25
For the zwitterionic form of mABA (mABA±), during the gas-phase geometry optimization of (mABA±)2 dimers we have observed inter-molecular H-transfer between the two monomers and the formation of (mABA)2 dimers. Therefore, to model the formation of (mABA±)2, the geometry optimization and frequency calculations were conducted in solution using the SMD solvation model, the B97-D density functional,35 and the 6-31++G(d,p) basis set. More accurate energetics were derived from single-point energy calculations at the SMD/M06-2X/aug-cc-pVDZ level of theory. The total free energies of mABA± and (mABA±)2 were then evaluated according to eqn (5) and (6).
Molecular systems like m-aminobenzoic acid and m-hydroxybenzoic can form complex mHBA/mHBA and mABA/mABA intermolecular interactions, i.e. XH/O and XH/π (X = O, N) hydrogen bonds and π–π interactions, all of which are important generally in polymorph formation. Therefore, in order to determine all the configurational minima of the dimers of TTA, mABA and mHBA, the starting structures for the geometry optimization procedure were obtained by chemical intuition, from the trajectories of ab initio molecular dynamics simulations (using the CP2K version 2.3.1636 and the PBE-D functional35) of (mABA)2, (mABA)(mABA±), (mABA±)2, and (mHBA)2 pairs. A simple algorithm was also developed to generate, from the structure of the lowest energy monomer, an arbitrary set of dimer structures.
Results and discussion
In this section we report the structure and stability of the self-associates of tetrolic acid, m-aminobenzoic acid and m-hydroxybenzoic acid in the gas-phase and different solvation environments.Tetrolic acid
Tetrolic acid is a simple carboxylic acid with two polymorphic forms: the metastable α-TTA form, which is based on a classic dimer motif and crystallizes from carbon tetrachloride or chloroform, and the stable β-TTA form, which is based on a catameric chain structure and can be recovered from alcoholic solutions.4 We have explored the configurational space of the (TTA)2 dimer by generating 60 starting geometries, and the free energies of dimerization of the “surviving” candidate structures in the gas-phase (ΔGoass) and in polar (water and methanol) and nonpolar (cyclohexane and chloroform) polarizable solvents (ΔG*ass) are reported in Table 2. All dimers are unstable with respect to the association of TTA molecules but c51_(TTA)2. This is the only dimer for which the association process is exoergonic in the gas phase (ΔG*ass = −4.52 kcal mol−1) and in the apolar solvents cyclohexane (ΔG*ass = −3.99 kcal mol−1) and chloroform (ΔG*ass = −0.64 kcal mol−1), but not in water and ethanol (ΔG*ass= 2.82 kcal mol−1). The structures of c51_(TTA)2 and of other representative (TTA)2 dimers are reported in Fig. 1. The species c51_(TTA)2 corresponds to the classic dimeric structural synthon found in the metastable α-TTA polymorph, which crystallizes from chloroform but not from alcoholic solutions. This result agrees with the experimental interpretation of the IR spectrum of tetrolic acid solutions, which indicates that in chloroform TTA molecules can form stable carboxylic acid dimers,8 and with previous classical MD simulations of tetrolic acid molecules in various solvents,15,17 which showed that the formation of the classic dimeric c51_(TTA)2 is favourable in chloroform and carbon tetrachloride but not in ethanol. In particular, despite the fact that continuum solvation models do not explicitly take into account the specific interactions between a solute and one or more first-shell solvent molecules, for example hydrogen bonding, it is important to notice that the free energies obtained in the present study for the formation of c51_(TTA)2 in chloroform (−0.64 kcal mol−1) and ethanol (+2.82 kcal mol−1) compare quite well with the free energy difference between the carboxylic acid dimer and the two fully solvated TTA monomers calculated by using the MD umbrella sampling method in chloroform (−1.1 kcal mol−1) and ethanol (+5.6 kcal mol−1).17 Therefore, in agreement with what was previous suggested, our calculations support the hypothesis that in chloroform there is a link between the solvent-dependent most thermodynamically stable dimer of TTA [the classic R22(8) carboxylic dimer synthon c51_(TTA)2] and the polymorph that crystallizes from solution (α-TTA). It is also interesting to notice that the second most stable dimer in ethanol, c8_(TTA)2 in Fig. 1 (ΔG*ass,EtOH = 4.24 kcal mol−1), corresponds to the structural motif found in the β-TTA crystal form.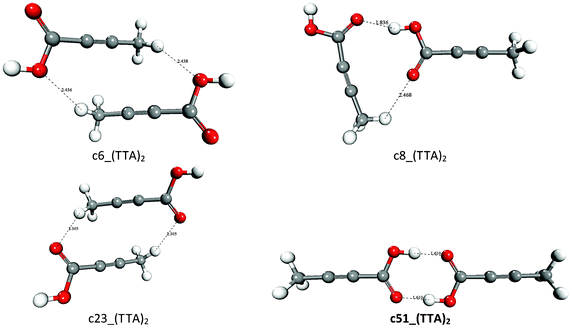 | ||
| Fig. 1 Optimized structures of representative (TTA)2 dimers. Distance in Å. | ||
| ΔEe,gas | ΔGoass | ΔG*ass | ||||
|---|---|---|---|---|---|---|
| Water | Ethanol | Cyclohexane | Chloroform | |||
| c1_(TTA)2 | −9.26 | 4.77 | 7.27 | 7.68 | 5.03 | 6.53 |
| c2_(TTA)2 | −6.28 | 7.86 | 8.68 | 9.27 | 8.22 | 9.00 |
| c3_(TTA)2 | −5.12 | 8.13 | 8.28 | 8.83 | 8.21 | 8.80 |
| c4_(TTA)2 | −6.28 | 5.97 | 6.76 | 7.34 | 6.31 | 7.09 |
| c5_(TTA)2 | −6.27 | 7.83 | 8.60 | 9.19 | 8.19 | 8.98 |
| c6_(TTA)2 | −8.17 | 2.61 | 3.97 | 4.29 | 2.94 | 3.86 |
| c8_(TTA)2 | −12.26 | 0.10 | 4.24 | 4.34 | 0.68 | 2.60 |
| c10_(TTA)2 | −6.33 | 7.71 | 9.89 | 10.25 | 7.87 | 9.25 |
| c12_(TTA)2 | −6.27 | 6.78 | 7.57 | 8.14 | 7.12 | 7.89 |
| c13_(TTA)2 | −6.34 | 7.75 | 9.93 | 10.26 | 7.90 | 9.26 |
| c14_(TTA)2 | −6.28 | 7.64 | 8.48 | 9.05 | 8.00 | 8.79 |
| c15_(TTA)2 | −6.28 | 6.56 | 7.33 | 7.89 | 6.89 | 7.67 |
| c16_(TTA)2 | −7.59 | 6.53 | 7.07 | 7.90 | 7.09 | 7.82 |
| c18_(TTA)2 | −6.86 | 6.96 | 7.01 | 7.52 | 7.34 | 7.86 |
| c23_(TTA)2 | −7.56 | 5.73 | 6.32 | 7.14 | 6.31 | 7.03 |
| c27_(TTA)2 | −7.59 | 5.76 | 6.30 | 7.13 | 6.31 | 7.03 |
| c31_(TTA)2 | −5.22 | 7.57 | 7.68 | 8.22 | 7.59 | 8.25 |
| c32_(TTA)2 | −6.28 | 6.09 | 6.87 | 7.43 | 6.42 | 7.19 |
| c33_(TTA)2 | −5.12 | 8.21 | 8.40 | 8.97 | 8.28 | 8.89 |
| c34_(TTA)2 | −9.25 | 4.11 | 6.73 | 7.67 | 4.38 | 6.23 |
| c35_(TTA)2 | −5.12 | 7.95 | 8.16 | 8.72 | 8.00 | 8.64 |
| c36_(TTA)2 | −6.28 | 6.50 | 7.24 | 7.87 | 6.86 | 7.66 |
| c37_(TTA)2 | −7.59 | 4.32 | 4.84 | 5.67 | 4.88 | 5.62 |
| c38_(TTA)2 | −9.25 | 4.12 | 6.65 | 6.98 | 4.35 | 5.79 |
| c39_(TTA)2 | −6.28 | 7.50 | 8.33 | 8.91 | 7.82 | 8.63 |
| c40_(TTA)2 | −5.12 | 7.84 | 7.97 | 8.52 | 7.88 | 8.46 |
| c41_(TTA)2 | −6.28 | 7.87 | 8.67 | 9.25 | 8.23 | 9.01 |
| c46_(TTA)2 | −6.34 | 7.58 | 9.60 | 9.94 | 7.69 | 9.04 |
| c48_(TTA)2 | −9.26 | 4.80 | 7.34 | 7.70 | 5.04 | 6.54 |
| c49_(TTA)2 | −6.34 | 7.07 | 9.20 | 9.55 | 7.22 | 8.58 |
| c50_(TTA)2 | −5.60 | 9.59 | 9.70 | 10.04 | 8.71 | 9.55 |
| c51_(TTA)2 | −16.63 | −4.52 | 2.82 | 2.82 | −3.99 | −0.64 |
| c53_(TTA)2 | −9.26 | 4.24 | 6.80 | 7.18 | 4.49 | 5.97 |
Starting from the carboxylic acid dimer c51, we have then computed the free energies for the formation of the trimers, (TTA)3, and tetramers, (TTA)4, of tetrolic acid in the gas-phase and chloroform. For the trimers, 60 candidate structures were generated using as building units the optimized gas-phase geometries of TTA and c51_(TTA)2. The free energies of association of these species have been computed according to the following association reaction:
| (TTA)2 + TTA → (TTA)3 | (7) |
The structures and free energies of the most stable trimers are reported in Fig. 2, whilst the free energies of reactions in the gas-phase and chloroform for the full set of generated trimers are listed in Table SI.1 of ESI.† The formation of a molecular aggregate (TTA)3 is favourable when the interaction occurs between the carboxylic groups of the tetrolic acid molecules [c10_(TTA)3, c37_(TTA)3 and c39_(TTA)3], but the most stable configuration of (TTA)3 is c60_(TTA)3, where the negatively charged sp hybridized carbon atoms of the TTA monomer interact with the positively charged hydrogen atoms of the hydroxyl groups of the dimer (Csp⋯H–O).
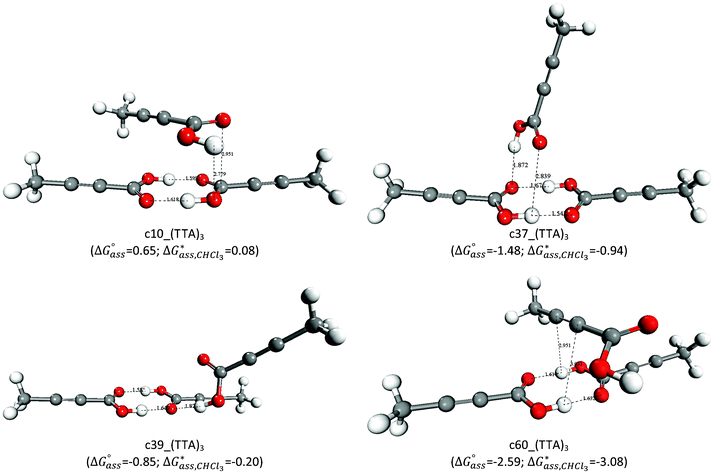 | ||
| Fig. 2 Optimized structures of representative (TTA)3 trimers and free energies of formation in the gas-phase and chloroform. Distances in Å and free energies in kcal mol−1. | ||
For the tetramers, starting from the structure of the dimer [c51_(TTA)2], we have generated and optimized 50 candidate structures, and the free energies of formation of (TTA)4 in the gas-phase and chloroform have been computed using the following reactions:
| (TTA)3 + TTA → (TTA)4 | (8) |
| (TTA)2 + (TTA)2 → (TTA)4 | (9) |
In chloroform, the free energies of reactions computed according to reaction (8) are positive for all tetramers (see Table SI.2 in the ESI†), whereas for reaction (9) we could identify two stable tetramers in the liquid-phase: c18_(TTA)4, ΔG*ass = −1.55 kcal mol−1, and c35_(TTA)4, ΔG*ass = −0.33 kcal mol−1) (see Table SI.3 in ESI†).
The results of the molecular association of TTA in chloroform are summarized in Fig. 3, where we have reported the structures and free energies of association in solution of the most stable dimer [c51_(TTA)2], trimer [c60_(TTA)3] and tetramer [c18_(TTA)4]. The results reported in Fig. 3 would suggest that the tetramerization of TTA molecules occurs from the self-association of the carboxylic acid dimers c51_(TTA)2 and not through the association of the TTA monomers and trimers. However, it is important to notice that the approach used in the present study to investigate the formation of trimers and tetramers excludes the possibility that trimers could also form from the association of solvated monomers:
| 3(TTA) → (TTA)3 | (10) |
| (TTA)2 + 2(TTA) → (TTA)4 | (11) |
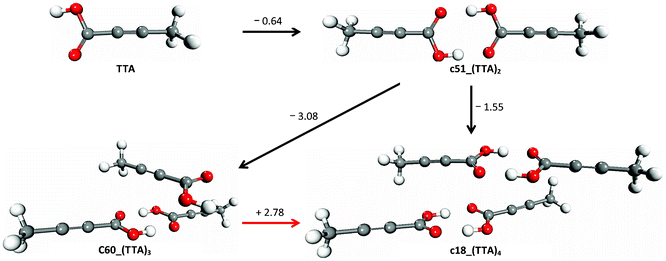 | ||
| Fig. 3 The most stable monomer, dimer, trimer and tetramer of TTA in chloroform. Structures optimised in the liquid-phase at the SMD/M06-2X/6-31++G(d,p) level of theory. Free energies in kcal mol−1. | ||
For example, chain-like trimers could become energetically competitive with the carboxylic acid dimer and the process of molecular-aggregation in solution could differ from the one outlined in Fig. 3. On the other hand, the configurational space that must be explored to simulate the formation of (TTA)3 and (TTA)4 according to eqn (10) and (11) is computationally very demanding for the full DFT-based approach employed in the present study.
m-Aminobenzoic acid
In the solution state m-aminobenzoic acid (mABA) can exists in both zwitterionic and non-zwitterionic forms. The nucleation is expected to be dependent on the distribution between non-zwitterionic and zwitterionic molecules, which is expressed by the equilibrium constant KZ = [mABA±]/[mABA]; the value of KZ has been found to be solvent-dependent.37 Also, this system exhibits abundant polymorphism, with five reported forms:38 in form I, III and IV the m-aminobenzoic acid exists in zwitterionic form, whereas in forms II and V the molecules are non-zwitterionic. Moreover, the nucleation of the mABA polymorphs depends chiefly on the solvent: in water and methanol the form I is obtained whereas form II crystallizes from acetonitrile.37In this study we have computed the formation of non-zwitterionic–non-zwitterionic [(mABA)2], non-zwitterionic–zwitterionic, [(mABA)(mABA±)], and zwitterionic–zwitterionic [(mABA±)2] dimers in the gas-phase and in water, methanol and acetonitrile. The dimerization free energies and structures of a representative set of non-zwitterionic dimers (mABA)2 are reported in Table 3 and Fig. 4, respectively. The free energies of formation of the full set of dimers generated to explore the configurational space of (mABA)2 are reported in Table SI.4 of ESI.† Only two species [c60_(mABA)2 and c61_(mABA)2] are stable in the gas- and liquid-phases (see Table 3) and, as shown in Fig. 4, these dimers correspond to the classic carboxylic acid dimer. On the other hand, the formation of (mABA)2 species where the monomers interact via other types of H-bonds [c3_(mABA)2, c41_(mABA)2, c48_(mABA)2], π–π interactions [c70_(mABA)2], or a combination of H-bonding and π–π interactions [c66_(mABA)2 and c71_(mABA)2], is endergonic in all solvation environments. The dimer c60_(mABA)2 corresponds to the carboxylic dimeric unit that has been found in form II of mABA,37 and its stability increases significantly on going from water (ΔG*solv = −0.35 kcal mol−1) and methanol (ΔG*solv = −0.06 kcal mol−1) to acetonitrile (ΔG*solv = −2.92 kcal mol−1), from where the polymorphic form II of mABA crystallizes. Therefore, similarly to what was found for tetrolic acid, the solvent controls the stability of the dimers in solution and, as suggested by the link hypothesis, the most stable dimer of mABA in acetonitrile [the carboxylic dimer synthon c60_(mABA)2] corresponds to the structural synthons packed in the polymorph that crystallizes from this solvent (form II).
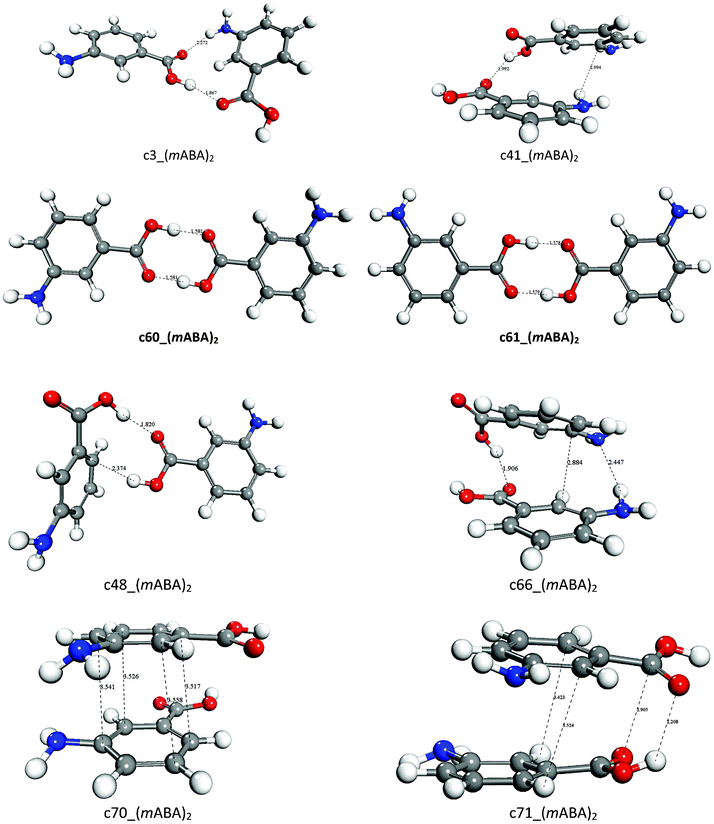 | ||
| Fig. 4 Optimized structures of representative (mABA)2 dimers. Distance in Å. | ||
| ΔEe,gas | ΔGoass | ΔG*ass | |||
|---|---|---|---|---|---|
| Water | Methanol | Acetonitrile | |||
| c3_(mABA)2 | −11.35 | −0.43 | 3.85 | 4.15 | 3.16 |
| c41_(mABA)2 | −11.14 | 0.70 | 2.27 | 3.16 | 3.59 |
| c48_(mABA)2 | −6.59 | 3.82 | 4.14 | 5.19 | 4.30 |
| c60_(mABA)2 | −17.79 | −6.68 | −0.35 | −0.06 | −2.92 |
| c61_(mABA)2 | −17.73 | −5.31 | 1.07 | 1.29 | −1.55 |
| c66_(mABA)2 | −8.75 | 4.71 | 4.60 | 5.63 | 2.41 |
| c70_(mABA)2 | −10.88 | 2.24 | 1.28 | 1.85 | 4.16 |
| c71_(mABA)2 | −10.80 | 1.99 | 2.08 | 2.68 | 3.98 |
In aqueous solutions the distribution of the non-zwitterionic and zwitterionic forms of mABA is close to unity.39 We have therefore computed the free energies for the formation of (mABA)(mABA±) dimers in water and, for comparison, also in methanol and acetonitrile, and the results are reported in Table SI.5 of ESI.† We have not been able to find values of KZ in acetonitrile, but in other aprotic solvents like dioxane and chloroform it has been shown that mABA is largely present in its non-ionic form.40 Notwithstanding, in acetonitrile the process of association of mABA and mABA± has been found to be endergonic, as for all generated dimers ΔG*solv > 0 (see Table SI.5 of ESI†), which implies that under the simulated conditions molecular associates of the type (mABA)(mABA±) are unlikely to be stable in this solvent. In water and methanol only one dimeric structure, c37_(mABA)(mABA±), has been found to be stable with respect to the association of mABA and mABA± molecules (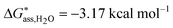 and
and  ). Its structure corresponds to a sandwich-like benzene dimer that allows the formation of a double H-bond (H2N+–H⋯NH2 and C–O−⋯HO–C) and π–π interactions between the mABA and mABA± molecules (see Fig. 5).
). Its structure corresponds to a sandwich-like benzene dimer that allows the formation of a double H-bond (H2N+–H⋯NH2 and C–O−⋯HO–C) and π–π interactions between the mABA and mABA± molecules (see Fig. 5).
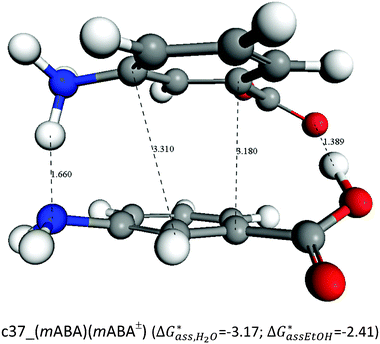 | ||
| Fig. 5 Optimized structure free energies of formation of the most stable (mABA)(mABA±) dimer in water and methanol. Distance in Å and free energies in kcal mol−1. | ||
Because in aqueous and alcoholic solutions the zwitterionic form of mABA is also likely to form molecular associates, we have computed the free energy of formation of (mABA±)2 dimers in water and methanol. The structures and free energies in water for a representative set of zwitterionic dimers (mABA±)2 are reported in Fig. 6, whilst the free energies of formation in water and methanol of the full set of dimers that have been generated to explore the configurational space of (mABA±)2 are reported in Tables SI.6 and SI.7 of ESI,† respectively. In water the most stable dimer, c30_(mABA±)2, is stabilized by a double H-bond (H2N+–H⋯−OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and π–π interactions between the two rings (see Fig. 6). When the monomers are not in an optimal position to form π–π interactions, then the stabilization free energy reduces to approximately −7 kcal mol−1 [c6_(mABA±)2, c9_(mABA±)2, c52_(mABA±)2 and c55_(mABA±)2], whereas when they can only form a single H-bond [c31_(mABA±)2, c32_(mABA±)2 and c34_(mABA±)2] the stabilization free energy is in the range of −1/−4 kcal mol−1. Similar results have been found for (mABA±)2 in methanol. Therefore, according to our results, the zwitterionic–zwitterionic dimers might be abundant in supersaturated aqueous and alcoholic solutions of m-aminobenzoic acid, a result that could be related to the abundant polymorphism exhibited by the zwitterionic form of mABA.38
O) and π–π interactions between the two rings (see Fig. 6). When the monomers are not in an optimal position to form π–π interactions, then the stabilization free energy reduces to approximately −7 kcal mol−1 [c6_(mABA±)2, c9_(mABA±)2, c52_(mABA±)2 and c55_(mABA±)2], whereas when they can only form a single H-bond [c31_(mABA±)2, c32_(mABA±)2 and c34_(mABA±)2] the stabilization free energy is in the range of −1/−4 kcal mol−1. Similar results have been found for (mABA±)2 in methanol. Therefore, according to our results, the zwitterionic–zwitterionic dimers might be abundant in supersaturated aqueous and alcoholic solutions of m-aminobenzoic acid, a result that could be related to the abundant polymorphism exhibited by the zwitterionic form of mABA.38
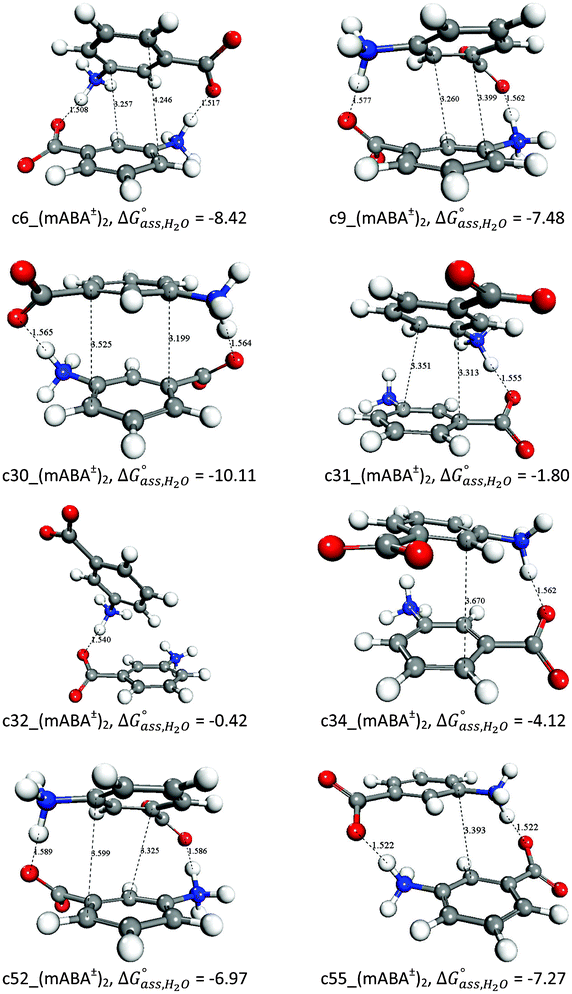 | ||
| Fig. 6 Optimized structures of representative zwitterionic–zwitterionic (mABA±)2 dimers and free energies of formation in water. Distances in Å and free energies in kcal mol−1. | ||
m-Hydroxybenzoic acid
m-Hydroxybenzoic acid (mHBA) crystallizes in two different polymorphic forms: the thermodynamically stable form I that contains the classic carboxylic dimer motif, and the metastable form II featuring intermolecular hydrogen bond chains between the COOH and OH groups of different molecules.41 Calculations at the M06-2X/aug-cc-pVDZ/6-31++G(d,p) level of theory indicated that mHBA exists mainly as four, almost iso-energetic, conformers, for which optimized geometries and relative energies are reported in Fig. 7. Another four stable forms were found by pointing the –OH of the carboxylic group towards the hydrogen of the benzene group, but their optimised structures were found to be 5.3–7.2 kcal mol−1 higher in energy than the lowest conformer, and therefore less statistically important than conformers I–IV. The geometries of conformers I–IV were then used to generate and optimize the structures of 140 (mHBA)2 dimers using the following combinations of mHBA conformers: conformer-I–conformer-I, conformer-II–conformer-II, conformer-III–conformer-III, conformer-VI–conformer-IV and conformer-II–conformer-III. The free energies of formation and structures for a representative set of (mHBA)2 dimers are reported in Table 4 and Fig. 8, respectively, whereas the free energies for the full set of candidate dimers that were generated to explore the configurational space of (mHBA)2 are reported in Table SI.8 of ESI.†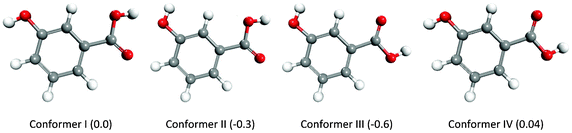 | ||
| Fig. 7 Optimized structures and relative energies (in kcal mol−1) of the conformers of mHBA as computed at the M06-2X/aug-cc-pVDZ/6-31++G(d,p) level of theory. | ||
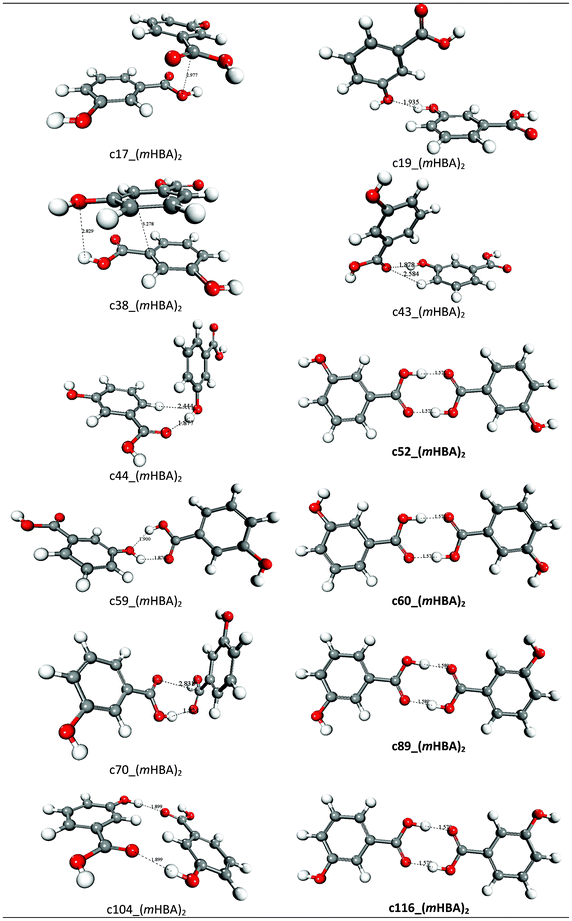 | ||
| Fig. 8 Optimized structures of representative (mHBA)2 dimers. Distances in Å. | ||
| ΔEe,gas | ΔGoass | ΔG*ass | ||||
|---|---|---|---|---|---|---|
| Water | Ethanol | Acetonitrile | Ethyl acetate | |||
| c17_(mHBA)2 | −6.26 | 4.63 | 1.75 | 2.66 | 4.36 | 4.71 |
| c19_(mHBA)2 | −7.09 | 3.59 | 4.06 | 4.51 | 4.19 | 3.74 |
| c38_(mHBA)2 | −8.39 | 3.53 | 0.71 | 1.83 | 3.62 | 3.86 |
| c42_(mHBA)2 | −9.63 | 0.59 | 2.70 | 3.15 | 2.41 | 1.55 |
| c44_(mHBA)2 | −9.64 | 1.62 | 3.78 | 4.18 | 3.38 | 2.54 |
| c52_(mHBA)2 | −17.32 | −6.07 | −0.01 | 0.08 | −2.40 | −3.95 |
| c59_(mHBA)2 | −11.76 | −0.66 | 4.67 | 4.91 | 3.95 | 2.21 |
| c60_(mHBA)2 | −18.03 | −6.71 | 0.01 | 0.17 | −2.20 | −4.12 |
| c70_(mHBA)2 | −9.01 | 2.30 | 3.92 | 7.39 | 4.07 | 3.16 |
| c89_(mHBA)2 | −17.61 | −4.43 | 1.91 | 2.12 | −0.31 | −2.04 |
| c104_(mHBA)2 | −15.31 | −2.64 | 5.04 | 5.26 | 2.96 | 1.13 |
| c116_(mHBA)2 | −17.51 | −6.36 | 0.32 | 0.47 | −2.03 | −3.86 |
Similarly to what was found for tetrolic acid and the non-zwitterionic form of m-aminobenzoic acid, the classic carboxylic acid dimers c52_(mHBA)2, c60_(mHBA)2, c89_(mHBA)2 and c116_(mHBA)2 (see Fig. 7) are the only stable species in acetonitrile and ethyl acetate, whereas in water and methanol the free energy of formation of these species is close to zero. In particular, the species c89_(mHBA)2 corresponds to the classic dimeric structural synthon found in the thermodynamically stable form I. The formation of other (mHBA)2 species, where the monomers interact via π–π interactions or single H-bonds between the COOH and OH groups, is endergonic in all solvation environments (see Tables 4 and SI.8 in ESI†).
Therefore if the “link hypothesis” is correct, then the polymorph containing the carboxylic dimer motif should preferentially crystallize from the solvent that mostly stabilizes the centrosymmetric carboxylic acid dimer c52_(mHBA)2, that is ethyl acetate (see Table 5). Svärd and Rasmuson have recently reported nucleation experiments of mHBA in several different pure solvents: in alcoholic solutions (ethanol and methanol) form I appeared to be favoured over form II; in water, acetonitrile and ethyl acetate the percentage of nucleations resulting in form II was more than 80%.42 The preferential nucleation from ethanol of form I, the polymorph containing the carboxylic dimer motif, partially agrees with the computed free energies of formation of carboxylic acid dimers in ethanol, which are close to zero and significantly lower than the free energies of other dimer structures (see Table 5). On the other hand, our theoretical prediction that in acetonitrile and ethyl acetate the carboxylic dimers are the most stable species in solution does not correlate with the preferential crystallization of form II from these solvents. Moreover, this result also contrasts to what was found for TTA and mABA, where the polymorph containing the carboxylic dimeric structural synthon nucleates from the solvent that mostly stabilizes the carboxylic acid dimer, chloroform and ethyl acetate, respectively. This suggests that there is not always a connection between the most thermodynamically stable molecular self-associate in a certain solvent and the polymorph that crystallizes from that particular solvent, as dictated by the “link hypothesis”. Therefore, this study suggests that it is not possible to predict the formation of a specific polymorph only from the knowledge of the most thermodynamically stable molecular self-associate in solution. The observed effect of the solvent on the free energy of formation of molecular associates, and consequently on the population statistics of molecular clusters in solution, is not the only mechanism dictating the outcome of a crystallization process. Other mechanisms, occurring for example at the crystal–liquid interface, could be also responsible for the preferential growth of a specific polymorph of mHBA over another.
| Level of theory | ΔEe,gas | ΔGoass | ΔG*ass | ||||
|---|---|---|---|---|---|---|---|
| Water | Ethanol | Acetonitrile | Ethyl acetate | ||||
| c38_(mHBA)2 | M06-2X/6-31+G(d,p) | −8.51 | 3.42 | 0.85 | 1.92 | 3.58 | 3.76 |
| M06-2X/6-31++G(d,p) | −8.26 | 3.43 | 0.85 | 1.93 | 3.59 | 3.77 | |
| M06-2X/aug-cc-pVDZ | −8.39 | 3.53 | 0.71 | 1.83 | 3.62 | 3.86 | |
| PBE/aug-cc-pVDZ | 0.50 | 12.82 | 9.45 | 10.52 | 12.23 | 12.48 | |
| PBE-D/aug-cc-pVDZ | −8.36 | 3.56 | 0.59 | 1.66 | 3.36 | 3.62 | |
| B97-D/aug-cc-pVDZ | −8.41 | 3.52 | 0.60 | 1.67 | 3.36 | 3.59 | |
| B3LYP/aug-cc-pVDZ | 3.75 | 15.68 | 12.82 | 13.89 | 15.57 | 15.79 | |
| c52_(mHBA)2 | M06-2X/6-31++G(d,p) | −18.40 | −7.15 | −0.90 | −0.66 | −3.09 | −4.79 |
| M06-2X/aug-cc-pVDZ | −17.32 | −6.07 | −0.01 | 0.08 | −2.40 | −3.95 | |
| PBE/aug-cc-pVDZ | −18.62 | −7.37 | −1.78 | −1.52 | −3.80 | −5.29 | |
| PBE-D/aug-cc-pVDZ | −20.91 | −9.66 | −4.07 | −3.81 | −6.09 | −7.58 | |
| B97-D/aug-cc-pVDZ | −17.91 | −6.36 | −0.67 | −0.45 | −2.79 | −4.28 | |
| B3LYP/aug-cc-pVDZ | −16.61 | −5.37 | 0.55 | −1.64 | −3.18 | ||
| c59_(mHBA)2 | M06-2X/aug-cc-pVDZ | −11.76 | −0.66 | 4.67 | 4.91 | 3.95 | 2.21 |
| PBE/aug-cc-pVDZ | −11.05 | −0.05 | 4.30 | 4.66 | 3.95 | 2.38 | |
| PBE-D/aug-cc-pVDZ | −13.59 | −2.49 | 1.87 | 2.22 | 1.52 | −0.06 | |
| B97-D/aug-cc-pVDZ | −11.49 | −0.49 | 3.87 | 4.21 | 3.48 | 1.92 | |
| B3LYP/aug-cc-pVDZ | −9.80 | 1.29 | 5.90 | 6.23 | 5.44 | 3.81 | |
In order to verify the accuracy of our calculations, the free energies of formation of one carboxylic acid dimer, c52_(mHBA)2, and those of the species c38_(mHBA)2 and c59_(mHBA)2 were evaluated using different basis sets [6-31+G(d,p), 6-31++G(d,p) and aug-cc-pVDZ] and density functionals (M06-2X, PBE, PBE-D, B97D and B3LYP). Species c38_(mHBA)2 was chosen because the mHBA molecules interact via π–π and hydrogen bonding in this dimer (see Fig. 8), and c59_(mHBA)2 was chosen because it contains the intermolecular H-bond motif found in the polymorphic form-II of mHBA. The results in Table 5 show that all methods agree in predicting the carboxylic acid dimer c52_(mHBA)2 as the most stable species in all solvation media. It is also important to point out that the gas phase interaction energies of the structure c38_(mHBA)2 computed using the meta-hybrid DFT method M06-2X and the generalized gradient DFT methods with dispersive correction, PBE-D and B97-D, differ by less than 0.3 kcal mol−1. The PBE and B3LYP stabilization energies computed on the optimised M06-2X geometries, however, are positive and more than 10 kcal mol−1 higher than the free energies obtained at the M06-2X level of theory. This is due to the neglecting dispersion at the PBE and B3LYP calculations. Moreover, starting from the M06-2X/6-31++G(d,p) optimized geometry of c38_(mHBA)2, we have re-optimized the structure at the PBE, PBE-D, B97D and B3LYP levels of theory (see Fig. 9). The structures obtained at the PBE-D and B97D levels are similar to the M06-2X optimised geometry. On the other hand, PBE generates a structure where the two monomers interact through H-bonds between the COOH and OH groups, whereas in the B3LYP structure the rings of the two mHBA monomers are more than 4.5 Å apart; this result shows that these functionals utterly fail to treat the stacking interaction.31,43
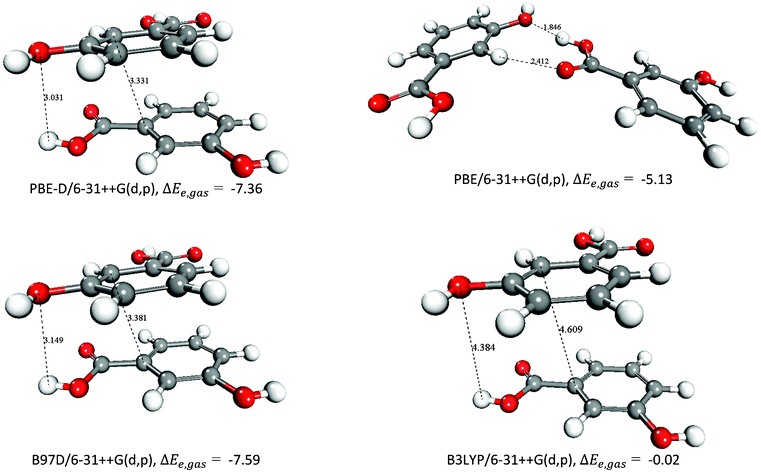 | ||
| Fig. 9 Structures of the c38_(mHBA)2 dimer and interaction energies as computed using different DFT methods. Distances in Å and gas-phase interaction energies in kcal mol−1. | ||
Conclusions
We have reported a computational investigation of the process of molecular self-association in different solvation environments of three selected monocarboxylic acids that crystallize in different polymorphic forms and exhibit selective crystallization depending on the solvent: tetrolic acid, m-aminobenzoic acid and m-hydroxybenzoic acid. The free energies of formation, both in the gas-phase and in solution (water and organic solvents), were computed at the DFT (M06-2X) level with the SMD polarizable model to simulate the solution environment. One of the aims of this study was to verify whether a link exists between the most stable molecular associates of TTA, mABA and mHBA in solution and the polymorph obtained from crystallization. Based on the results of our calculations and comparison with nucleation experiments, we conclude the following.For all three selected monocarboxylic acids the most stable dimer in solution is the classic carboxylic acid dimer, and its stability decreases on going from nonpolar solvents, such as chloroform or acetonitrile, to polar environments, like water and alcoholic solvents.
For TTA and mABA our calculations support the hypothesis that there is a connection between the carboxylic dimer synthon in solution and the structural synthon packed in the metastable α-TTA polymorph, which crystallizes from chloroform, and in the crystal form-II of mABA, which crystallizes from acetonitrile, in agreement with the results proposed by the “link” hypothesis. However, for mHBA, the stabilization of the centrosymmetric carboxylic acid dimer in ethyl acetate and acetonitrile does not correspond with recent experimental observations that the crystal form containing the classic carboxylic dimer motif (form I) is not the dominant polymorph that crystallizes from these solvents.42
This study shows that for carboxylic acids the structure of the most stable species in solution (the classic carboxylic acid dimer) is not always linked to the structural synthon of the polymorph that crystallizes from solution. Consequently, the formation of a specific polymorph is not only dependent on the population statistics of molecular clusters in solution. Other factors, such as the interactions occurring at the crystal–liquid interfaces of the polymorphs of mHBA, could be important in directing the polymorphic outcome of a crystallization process.
Trimers of TTA are stable in chloroform, but the formation of tetramers occurs from the self-association of the carboxylic acid dimers and not through the association of the TTA monomers and trimers. However, we have not considered the formation of trimers and tetramers in terms of the association of solvated monomers, as the configurational space to be explored would be too large and computationally demanding for the DFT-based approach employed in the present study. Therefore, we cannot exclude that the formation of trimers and tetramers could also occur through other alternative molecular-aggregation pathways.
The free energy formation of the ionized forms of the mABA dimer (non-zwitterionic–zwitterionic and zwitterionic–zwitterionic) has been evaluated and, according to our results, zwitterionic–zwitterionic dimers would be abundant in supersaturated aqueous and alcoholic solutions of m-aminobenzoic acid.
Acknowledgements
The author thanks the U.K.'s Royal Society for the award of a Royal Society Industry Fellowship. Computer resources on the UK ‘HECToR’ services were provided via the U.K.'s HPC Materials Chemistry Consortium (EPSRC grant no. EP/D504872). Dr Simon Black (AstraZeneca) is thanked for useful discussions.References
- J. O. Henck, U. J. Griesser and A. Burger, Pharm. Ind., 1997, 59, 165–169 Search PubMed.
- H. W. Sollinger, W. B. Vernon, A. M. Dalessandro, M. Kalayoglu, R. J. Stratta and F. O. Belzer, Surgery, 1989, 106, 685–691 CAS.
- M. Lahav and L. Leiserowitz, Chem. Eng. Sci., 2001, 56, 2245–2253 CrossRef CAS.
- S. Parveen, R. J. Davey, G. Dent and R. G. Pritchard, Chem. Commun., 2005, 1531–1533 RSC.
- R. J. Davey, N. Blagden, S. Righini, H. Alison, M. J. Quayle and S. Fuller, Cryst. Growth Des., 2001, 1, 59–65 CAS.
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS.
- R. J. Davey, G. Dent, R. K. Mughal and S. Parveen, Cryst. Growth Des., 2006, 6, 1788–1796 CAS.
- J. Chen and B. L. Trout, J. Phys. Chem. B, 2010, 114, 13764–13772 CrossRef CAS.
- R. J. Davey, N. Blagden, G. D. Potts and R. Docherty, J. Am. Chem. Soc., 1997, 119, 1767–1772 CrossRef CAS.
- R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS.
- S. Hamad, C. E. Hughes, C. R. A. Catlow and K. D. M. Harris, J. Phys. Chem. B, 2008, 112, 7280–7288 CrossRef CAS.
- S. Hamad, C. Moon, C. R. A. Catlow, A. T. Hulme and S. L. Price, J. Phys. Chem. B, 2006, 110, 3323–3329 CrossRef CAS.
- E. Khurana, S. O. Nielsen, B. Ensing and M. L. Klein, J. Phys. Chem. B, 2006, 110, 18965–18972 CrossRef CAS.
- A. Gavezzotti, G. Filippini, J. Kroon, B. P. van Eijck and P. Klewinghaus, Chem.–Eur. J., 1997, 3, 893–899 CrossRef CAS.
- Y. Yani, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2012, 12, 4771–4778 CAS.
- J. Chen and B. L. Trout, J. Phys. Chem. B, 2008, 112, 7794–7802 CrossRef CAS.
- L. J. Smith, H. J. C. Berendsen and W. F. van Gunsteren, J. Phys. Chem. B, 2004, 108, 1065–1071 CrossRef CAS.
- H. Tanaka, K. Nakanishi and H. Touhara, J. Chem. Phys., 1985, 82, 5184–5191 CrossRef CAS.
- S. Hamad and C. R. A. Catlow, CrystEngComm, 2011, 13, 4391–4399 RSC.
- F. Ramondo, L. Bencivenni, R. Caminiti, A. Pieretti and L. Gontrani, Phys. Chem. Chem. Phys., 2007, 9, 2206–2215 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 CrossRef.
- J. Klimeš and A. Michaelides, J. Chem. Phys., 2012, 137, 120901–120912 CrossRef.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2011, 13, 10908–10922 RSC.
- J. M. Ho, A. Klamt and M. L. Coote, J. Phys. Chem. A, 2010, 114, 13442–13444 CrossRef CAS.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.2), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 1849–1868 CrossRef CAS.
- J. A. Plumley and J. J. Dannenberg, J. Comput. Chem., 2011, 32, 1519–1527 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 2447–2464 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 4538–4543 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- J. Vandevondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- M. Svard, F. L. Nordstrom, T. Jasnobulka and A. C. Rasmuson, Cryst. Growth Des., 2010, 10, 195–204 CAS.
- P. A. Williams, C. E. Hughes, G. K. Lim, B. M. Kariuki and K. D. M. Harris, Cryst. Growth Des., 2012, 12, 3104–3113 CAS.
- W. D. Kumler, J. Org. Chem., 1955, 20, 700–706 CrossRef CAS.
- L. Gopal, C. l. Jose and A. B. Biswas, Spectrochim. Acta, Part A, 1967, 23A, 519–542 Search PubMed.
- F. L. Nordstrom and A. C. Rasmuson, Eur. J. Pharm. Sci., 2006, 28, 377–384 CrossRef.
- M. Svard and A. C. Rasmuson, Cryst. Growth Des., 2013, 13, 1140–1152 Search PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996–2000 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ce40539g |
| This journal is © The Royal Society of Chemistry 2013 |