 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards ab initio screening of co-crystal formation through lattice energy calculations and crystal structure prediction of nicotinamide, isonicotinamide, picolinamide and paracetamol multi-component crystals†
H. C. Stephen Chanab, John Kendricka, Marcus A. Neumannc and Frank J. J. Leusen*a
aSchool of Life Sciences, University of Bradford, Richmond Road, Bradford, BD7 1DP, UK. E-mail: f.j.j.leusen@bradford.ac.uk; Tel: +44 (0)1274 236144
bNovartis Institutes for Biomedical Research, Postfach, CH-4002 Basel, Switzerland
cAvant-garde Materials Simulation (Deutschland) GmbH, Merzhauser Strasse 177, D-79100 Freiburg, Germany
First published on 8th March 2013
Abstract
Co-crystallisation of a drug with another molecule to form a new crystalline material is an appealing route to enhance physical properties. Despite mounting research effort, there is still considerable uncertainty whether a given co-crystal will form. Previous attempts to use lattice energy calculations to investigate whether a potential co-crystal is thermodynamically more stable than its pure co-former crystals have been inconclusive. In the present study, dispersion-corrected density functional theory is used to minimise the lattice energies of all known co-crystals and salts of nicotinamide, isonicotinamide and picolinamide, and their corresponding neutral co-formers (excluding any organometallic compounds). Out of the resulting 102 co-crystals and salts, 99 (97%) are found to be more stable than their corresponding co-formers. In addition, full crystal structure prediction studies show that two paracetamol co-crystals are very unstable in comparison to their co-formers, thus explaining why these co-crystals have not been observed experimentally. These results demonstrate that a simple yet accurate thermodynamic approach can predict reliably whether a co-crystal can be formed.
Introduction
Research in co-crystals has seen dramatic growth in recent years, mainly driven by their pharmaceutical applications where properties such as solubility, hardness or tabletting1,2 are of paramount importance. In addition, co-crystals offer the potential to address intellectual property issues and create patent opportunities in pharmaceutical and other industries. The growth in research activity is illustrated by the number of entries for co-crystals in the Cambridge Structural Database.3 The number of organic co-crystals is 10 times higher in 2007 than in 1988 and 2.5 times higher than in 1997.4 Many experimental techniques can produce co-crystals5 and there have also been previous attempts to use lattice energy calculations to investigate whether a potential co-crystal is thermodynamically more stable than its pure co-former crystals.6–8 However, there is still considerable uncertainty as to whether a given co-crystal will form and questions still remain as to the thermodynamic or kinetic nature of (co-)crystallisation.Significant advances have been achieved recently in the accuracy of lattice energy calculations applied to the organic solid state through the use of a dispersion-corrected density functional theory (DFT-D) approach.9–13 Since a major issue with co-crystals is their stability, which is often not sufficient for pharmaceutical formulations,5 it is important to establish whether state-of-the-art DFT-D lattice energy calculation tools are accurate enough to determine the stability of co-crystals (and salts) relative to their solid state co-formers. 102 co-crystals and their co-formers are investigated in this study using an approach based solely on lattice energy, i.e., temperature, pressure, solvent effects and other kinetic factors are not considered in the calculations.
Consider the following reaction,
| xA(solid) + yB(solid) → AxBy(solid) + ΔH |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y and ΔH is the enthalpy change in the process. Furthermore,
:y and ΔH is the enthalpy change in the process. Furthermore,| ΔH ≈ EAB − (xEA + yEB) |
The results obtained by previous CSP studies on multi-component crystal systems show that the accuracy of the simulated potential energy surface and the search for all plausible structures becomes profoundly demanding for co-crystal structures.6–8 Indeed a CSP study on the pyridine molecule14 indicated that the number of crystal structures in a given energy window grows exponentially with Z′. Having successfully predicted the experimental structures of a 1:1 co-crystal in the 2007 CSP blind test15 and a molecular salt in the 2010 CSP blind test16 (as the first and third of the three submitted structures respectively), the DFT-D approach9 is applied in the second part of this work to investigate whether it is possible to predict a negative co-crystallisation result. To this end, two paracetamol co-crystals have been selected that, despite considerable efforts, have not been observed experimentally.
For the purpose of this study, all non-organometallic, anhydrous co-crystals and salts containing nicotinamide, isonicotinamide and picolinamide were selected for which full crystal structures of co-crystals and their co-formers are available, either from the Cambridge Structural Database (CSD)3 version 5.32 released in November 2011 or from the Inorganic Crystal Structure Database.17 Nicotinamide (vitamin B6) and isonicotinamide (see Fig. 1) have attracted considerable interest in co-crystallisation research. The former has been used as a co-crystal former of pharmaceuticals such as carbamazepine, ibuprofen and celecoxib. The latter has been studied extensively for the effects of intermolecular interactions on the formation of co-crystals.18,19 The co-crystal and salts of picolinamide, a structural isomer of nicotinamide, are also included in this study. The lattice energies of all reported polymorphs of the co-crystals, salts, and their single component co-formers were minimised with respect to atomic coordinates and unit cell dimensions using a DFT-D method implemented in the GRACE software package.20 DFT calculations were performed using VASP21,22 with the PW91 functional (see the ESI† for further details of the methodology).
 | ||
| Fig. 1 Molecular structures of a) nicotinamide, b) isonicotinamide, c) picolinamide, d) paracetamol, e) benzoic acid and f) anthranilic acid. | ||
An ab initio screening method of co-crystal formation also requires the ability to predict a negative result, i.e., the prediction that a co-crystal is less stable than its co-formers and can therefore not be crystallised. To investigate this aspect, the same DFT-D methodology, in conjunction with state-of-the-art CSP tools described previously,10–13 was applied to two paracetamol co-crystals which have not been observed experimentally. Paracetamol (see Fig. 1) is a widely used painkiller and anti-pyretic with at least three reported polymorphs (CSD codes: HXACAN01, HXACAN08 and HXACAN29). Anthranilic acid and benzoic acid (see Fig. 1) did not form any co-crystals with paracetamol in Kofler fusion tests and in solvent co-crystallisation experiments.23 These findings are unexpected because paracetamol is known to form co-crystals with oxalic acid, citric acid and 2,4-pyridinedicarboxylic acid (CSD codes: LUJTAM, AMUBAM and SUTVAF respectively). To investigate the performance of our ab initio screening method for co-crystal formation on these two negative cases, full CSP studies of the component molecules and the 1:1 paracetamol:acid co-crystals were conducted.
Results and discussion
The root-mean-square deviation (RMSD) of the non-hydrogen atoms between the fully optimised and the experimental structures of the co-former polymorphs provides a quantitative measure of the similarity for each pair of structures. Two crystals (out of 124 structures) show deviations larger than expected (>0.3 Å), indicating either a problem with the DFT-D method or a poor experimental structure. Form II rac-ibuprofen has a poor experimental molecular geometry, resulting in a RMSD of 0.50 Å after optimisation. The O–H bond of the carboxylic acid in the experimental structure of the orthorhombic form (Pcan) of diclofenac is exceptionally long (1.47 Å), and was shortened prior to optimisation. The diclofenac molecules are significantly displaced on optimisation to facilitate the dimerisation of the carboxylic acid groups, resulting in a RMSD of 0.69 Å. For the other 122 co-former structures, the RMSDs are smaller than 0.30 Å, which indicates that the geometric DFT-D results are in agreement with experiment and that these results can be used with confidence. The ESI† contains a detailed comparison of calculated and measured relative stabilities of co-former polymorphs along with a CIF file containing all the energy minimised crystal structures.Regarding the types of structures considered in this study, the co-formers form a total of 34 co-crystals and 10 salts with nicotinamide; 47 co-crystals, 3 hybrid salt co-crystals and 6 salts with isonicotinamide; and 1 co-crystal and 2 salts with picolinamide. Their stabilities were calculated with the DFT-D method relative to the sum of the co-former lattice energies (see Tables S1 and S2, ESI†). For polymorphic co-formers, the lattice energy of the most stable polymorph was used in the stability calculation. Out of the 102 co-crystals and salts, 99 are found to be more stable than their corresponding pure crystal components, i.e., considering only the thermodynamic stability of these compounds, calculated by accurate quantum mechanics and ignoring kinetics and other effects, these calculations successfully predict the co-crystal or salt to be more stable than their co-formers with a success rate of more than 97%. The results are graphically presented in Fig. 2, where the stabilities of the co-crystals and salts relative to their co-formers are shown as a histogram; see ESI† for details. The ESI† also contains CIF files with all the energy minimised co-crystal and salt crystal structures.
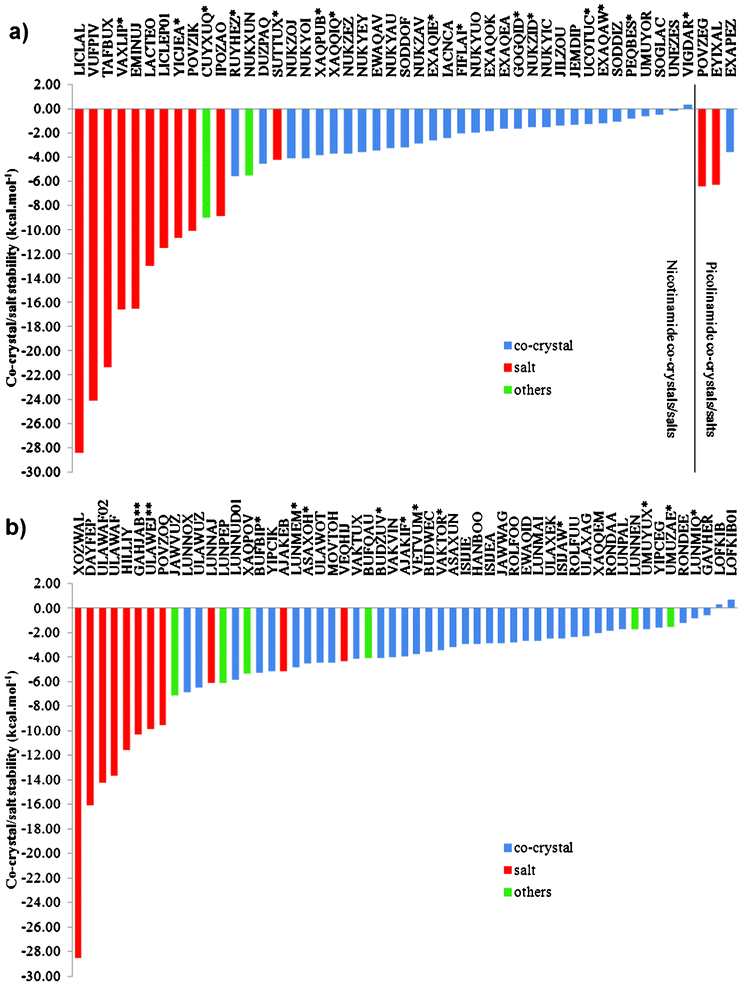 | ||
| Fig. 2 DFT-D calculated stabilities of a) nicotinamide and picolinamide co-crystals and salts and b) isonicotinamide co-crystals and salts. The structures are identified by their CSD reference codes. Blue indicates an optimised co-crystal structure. Red indicates that the optimised structure is a salt. Green indicates a hybrid structure. * and ** At least one of the experimental structures was modified prior to optimisation. | ||
The only co-crystals found to be less stable than their co-formers are celecoxib:nicotinamide and the two polymorphs of carbamazepine:isonicotinamide. The minimised lattice energy of the 1:1 celecoxib:nicotinamide co-crystal structure reported in the CSD is 1.82 kcal mol−1 higher than the energy of its components. This large, apparent instability may be caused in part by an incorrect experimental structure for the co-crystal as indicated by its exceptionally large RMSD (1.71 Å). A detailed investigation of the experimental structure revealed several irregularities. Since the refinement of the co-crystal structure relied heavily on powder X-ray diffraction data,24 it is possible that some nitrogen and oxygen atoms were interchanged, as they have similar atomic weight. With this in mind, 23 additional variations of the celecoxib:nicotinamide co-crystal structure were created starting with the packing reported in the CSD but introducing different conformations of celecoxib and nicotinamide (see ESI† for details). These alternative structures were optimised with the DFT-D method. The most stable structure facilitates intermolecular hydrogen bonding by a 180° rotation of the amide group in nicotinamide and a 120° anti-clockwise rotation (viewed along the S–C bond) of the sulfonamide group in celecoxib (see Fig. 3). The sulfonamide S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths (1.455 and 1.458 Å) in this new structure are more realistic than those in the reported structure (1.596 and 1.405 Å). Based on these geometric considerations and the lattice energy results (see Table S3, ESI†), we postulate that the true celecoxib:nicotinamide co-crystal structure corresponds to the optimised structure shown in Fig. 3b, except that the trifluoromethyl group is disordered. However, this co-crystal structure is still 0.36 kcal mol−1 less stable than its single component crystals. This small energy difference may be, at least in part, due to the stabilising effects of the disordered trifluoromethyl group, which are not taken into account in the lattice energy calculations. The proposed crystal structure of the celecoxib:nicotinamide co-crystal is provided in the ESI.†
O bond lengths (1.455 and 1.458 Å) in this new structure are more realistic than those in the reported structure (1.596 and 1.405 Å). Based on these geometric considerations and the lattice energy results (see Table S3, ESI†), we postulate that the true celecoxib:nicotinamide co-crystal structure corresponds to the optimised structure shown in Fig. 3b, except that the trifluoromethyl group is disordered. However, this co-crystal structure is still 0.36 kcal mol−1 less stable than its single component crystals. This small energy difference may be, at least in part, due to the stabilising effects of the disordered trifluoromethyl group, which are not taken into account in the lattice energy calculations. The proposed crystal structure of the celecoxib:nicotinamide co-crystal is provided in the ESI.†
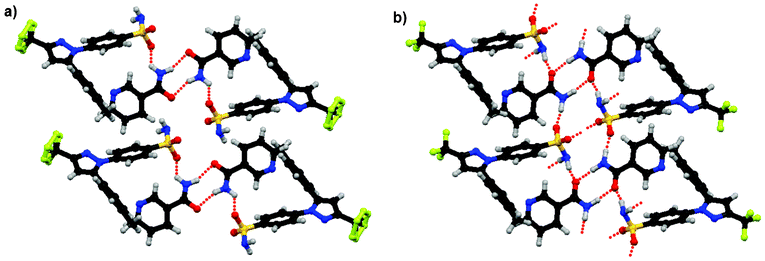 | ||
| Fig. 3 Molecular conformations and intermolecular packing in a) the reported celecoxib:nicotinamide co-crystal24 and b) the most stable optimised structure in this study. Dotted lines indicate hydrogen bonds. | ||
The minimised lattice energies of the two polymorphs of carbamazepine:isonicotinamide, forms I and II, are respectively 0.32 and 0.67 kcal mol−1 higher than the total lattice energy of pure carbamazepine and isonicotinamide. The calculated stabilities of the co-crystal polymorphs are consistent with the observations in solvent crystallisation experiments that the less stable form II first appears in the liquor and later transforms into the more stable form I co-crystal.25 It is unclear why the carbamazepine:isonicotinamide polymorphs are calculated to be less stable than their pure co-formers. Possible reasons include kinetic effects, although the experiments were conducted under standard conditions (co-crystallisation took place from pure crystalline components in ethanol solution at 25 °C), or some unknown inadequacy of the DFT-D method in treating this particular system.
The stabilities in Fig. 2 show an interesting distribution over co-crystals, salts and hybrid salt co-crystals structures. Note that optimisation of the experimental structures using the DFT-D method can change the nature of the crystals. Six (CSD codes AJAKEB, LUNNAJ, SUTTUX, ULAWAF, ULAWAF02, ULAWEJ) out of the 24 optimised salts were initially co-crystals or structures with proton bridges. Seven (BUFQAU, CUYXUQ, LUNNEN, LUNPEP, NUKXUN, UMUZAE, XAQPOV) out of the eight optimised ‘hybrid’ structures were co-crystals before optimisation. In all cases a proton from the acid moiety of the co-former migrates to the pyridine nitrogen of nicotinamide or isonicotinamide. The remaining 18 DFT-D optimised salts show no tendency to form neutral co-crystals. The average stability of the 24 DFT-D optimised salts relative to their co-formers is −12.81 kcal mol−1. For the remaining 70 co-crystals (excluding eight hybrid structures), the average relative lattice energy difference is −2.75 kcal mol−1. Hence, according to the DFT-D method, when salts form, they are considerably more stable than a co-crystal. This can be rationalised by considering that the strength of the hydrogen bond in a co-crystal is at most about 8 kcal mol−1, but the potential strength of the salt bridge, if it can be formed, is significantly greater. Some caution should be exercised in interpreting the relative stabilities of the co-crystal and its related salt as the DFT-D method, which has proved itself capable of predicting accurate relative lattice energetics for crystals with the same protonation state, has not been so thoroughly tested for cases where the protonation state can change.
The CSP results of paracetamol using the GRACE software have been published previously.26 The rank 1, 3 and 6 DFT-D predictions corresponded to the experimental polymorphs, form I, II and III respectively. The predicted order of stability was consistent with the experimental findings.26 Hence, in the current work, the minimised DFT-D lattice energy of form I was selected for calculating the co-crystal stabilities (see Table 1). Tailor-made force fields (TMFF)27 were parameterised for anthranilic acid and benzoic acid using reference data sets generated by the DFT-D method.9 The TMFF enables fast and fairly accurate calculations of lattice energies and atomic forces in the structure-generation step. All structures within a given energy window with one independent molecule in the asymmetric unit were generated in 230 space groups using a Monte Carlo parallel tempering search algorithm. These structures were considered for further optimisation by the DFT-D method. Note that the form I polymorph of anthranilic acid (CSD code: AMBACO07) contains a zwitterion and a neutral molecule in the asymmetric unit, and that the interchange of the ionisation states cannot be handled by a single TMFF. As the CSP was restricted to structures with one independent molecule in the asymmetric unit, this experimental structure could not have been predicted. Therefore, the experimental structure of form I anthranilic acid was optimised by the DFT-D method and included in the calculation of co-crystal stability. For each 1:1 paracetamol:acid co-crystal, the paracetamol TMFF from the earlier study26 was combined with the anthranilic acid or benzoic acid TMFF. Additional rigid-molecule minimisation data sets were generated by the DFT-D method to parameterise the van der Waals interactions between the component molecules. In the structure generation step, the asymmetric unit consisted of one independent paracetamol molecule and one independent acid molecule. The important CSP parameters are summarised in Table 2. The DFT-D minimised crystal structures found by CSP in this work are provided as a CIF file in the ESI.† A summary of the low-energy predicted structures is provided in Table 3. Crystal structure similarity was studied using the Compack algorithm28 in the Mercury software (version 2.3)29 which overlaid clusters of 16 molecules from the experimental and the DFT-D predicted crystal structures. The root-mean-square deviations of non-hydrogen atoms were calculated.
| Crystal structurea | Space group | Unit cell parametersb | EDFT-Dc [kcal mol−1] | Density [g cm−3] | RMSDd [Å] | |||
|---|---|---|---|---|---|---|---|---|
| a [Å] | b [Å] | c [Å] | β [°] | |||||
| a The experimental structures are shown using their entry codes in the Cambridge Structural Database (CSD).3b Angles α and γ are 90° due to symmetry.c The numbers in brackets are the relative energies from the most stable DFT-D optimised structure.d RMSD is the root-mean-square deviation of the non-hydrogen atoms between the experimental and the DFT-D optimised structures, calculated with the Compack algorithm28 as implemented in Mercury 2.329 using overlaid clusters of 16 molecules. | ||||||||
| Paracetamol | ||||||||
| HXACAN01 (Form I) | P21/a | 12.93 | 9.40 | 7.10 | 115.9 | 1.296 | ||
| DFT-D optimised | P21/a | 12.81 | 9.15 | 6.97 | 113.8 | −2986.38 | 1.344 | 0.193 |
| HXACAN08 (Form II) | Pbca | 17.17 | 11.78 | 7.21 | 90.0 | 1.377 | ||
| DFT-D optimised | Pbca | 17.22 | 11.59 | 7.31 | 90.0 | (+0.17) | 1.377 | 0.108 |
| HXACAN29 (Form III) | Pca21 | 11.84 | 8.56 | 14.82 | 90.0 | 1.418 | ||
| DFT-D optimised | Pca21 | 11.63 | 8.59 | 14.61 | 90.0 | (+0.32) | 1.376 | 0.182 |
:1 paracetamol:anthranilic acid and 1:1 paracetamol:benzoic acid
| Molecule | σ [kcal mol−1]a | Energy window used to select structures for re-ranking [σ] | Number of structures considered for re-ranking | Number of structures optimised by the DFT-D method |
|---|---|---|---|---|
| a σ is the standard deviation in energy comparing the TMFF and DFT-D results. | ||||
| Anthranilic acid | 0.69 | 3.0 | 165 | 76 |
| Benzoic acid | 0.59 | 5.1 | 768 | 48 |
| 1:1 Paracetamol:anthranilic acid | 1.70 | 3.0 | 692 | 138 |
| 1:1 Paracetamol:benzoic acid | 1.02 | 4.1 | 2827 | 1254 |
:1 paracetamol:anthranilic acid and 1:1 paracetamol:benzoic acid and the known experimental unit cell parameters
| Crystal structurea | Space group | Unit cell parametersb | EDFT-Dc [kcal mol−1] | Density [g cm−3] | RMSDd [Å] | |||
|---|---|---|---|---|---|---|---|---|
| a [Å] | b [Å] | c [Å] | β [°] | |||||
| Anthranilic acid | ||||||||
| AMBACO07 (Form I) | P21cn | 12.86 | 10.79 | 9.31 | 90.0 | |||
| DFT-D optimised Form I | P21cn | 12.90 | 10.77 | 9.20 | 90.0 | −2608.61 | 1.426 | 0.065 |
| AMBACO05 (Form II) | Pbca | 15.99 | 11.62 | 7.16 | 90.0 | |||
| CSP rank 1 | Pbca | 15.96 | 11.52 | 7.09 | 90.0 | (+0.74) | 1.397 | 0.071 |
| AMBACO08 (Form III) | P21/c | 6.54 | 15.35 | 7.09 | 112.6 | |||
| CSP rank 2 | P21/c | 6.44 | 15.35 | 6.95 | 112.1 | (+1.09) | 1.431 | 0.104 |
| CSP rank 3 | P21/c | 5.12 | 12.45 | 11.05 | 109.9 | (+1.26) | 1.377 | |
| CSP rank 4 | P21/c | 5.53 | 4.96 | 23.30 | 98.9 | (+1.30) | 1.442 | |
| CSP rank 5 | Pccn | 11.73 | 16.00 | 7.05 | 90.0 | (+1.44) | 1.378 | |
| CSP rank 6 | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | 7.06 | 6.96 | 7.49 | 105.0 | (+1.44) | 1.408 | |
| 82.5 | ||||||||
| 69.8 | ||||||||
| CSP rank 7 | P21/c | 5.06 | 5.27 | 23.88 | 93.8 | (+1.49) | 1.434 | |
| CSP rank 8 | C2/c | 16.17 | 7.05 | 13.15 | 59.8 | (+1.56) | 1.406 | |
| CSP rank 9 | P21/c | 7.21 | 11.32 | 8.07 | 74.2 | (+1.57) | 1.436 | |
| CSP rank 10 | P21/c | 8.15 | 4.06 | 19.96 | 97.3 | (+1.61) | 1.390 | |
| Benzoic acid | ||||||||
| BENZAC07 | P21/c | 5.39 | 5.00 | 21.69 | 98.5 | |||
| CSP rank 1 (2) | P21/c | 5.44 | 5.04 | 21.84 | 98.2 | −2317.15 | 1.368 | 0.081 |
| CSP rank 2 (1) | P21/c | 5.50 | 5.06 | 21.63 | 98.3 | (+0.06) | 1.363 | 0.116 |
| CSP rank 3 (4) | P21/c | 5.20 | 5.35 | 21.26 | 87.2 | (+0.08) | 1.373 | |
| CSP rank 4 (3) | P21/c | 5.22 | 5.41 | 21.11 | 87.0 | (+0.15) | 1.363 | |
| CSP rank 5 (10) | P21/c | 6.34 | 3.89 | 25.03 | 105.1 | (+0.36) | 1.360 | |
| CSP rank 6 | P21/c | 8.82 | 5.07 | 13.63 | 86.8 | (+0.43) | 1.332 | |
| CSP rank 7 (8) | P21/c | 6.02 | 16.19 | 6.76 | 112.3 | (+0.44) | 1.331 | |
| CSP rank 8 (7) | P21/c | 5.80 | 16.50 | 6.86 | 111.1 | (+0.48) | 1.324 | |
| CSP rank 9 | P21/c | 7.89 | 6.38 | 12.77 | 73.4 | (+0.50) | 1.316 | |
| CSP rank 10 (5) | P21/c | 6.38 | 3.92 | 24.83 | 104.9 | (+0.50) | 1.352 | |
| Crystal structurea | Space group | Unit cell parametersb | EDFT-Dc [kcal mol−1] | Density [g cm−3] | HBe | |||
|---|---|---|---|---|---|---|---|---|
| a [Å] | b [Å] | c [Å] | β [°] | |||||
| a The experimental structures are shown using their entry codes in the Cambridge Structural Database (CSD).3 For benzoic acid, the acidic proton in the experimental structure is disordered over site1 and site2 with occupancies 0.87:0.13 at 20 K. The number in brackets is the DFT-D ranking of the isostructural partner.b Unless otherwise specified, angles α and γ are 90° by symmetry.c The numbers in brackets are the relative energies from the most stable DFT-D predicted/optimised structures.d RMSD is the root-mean-square deviation of the non-hydrogen atoms between the experimental and the DFT-D predicted/optimised structures, calculated with the Compack algorithm28 as implemented in Mercury 2.329 using overlaid clusters of 16 molecules.e HB stands for hydrogen-bond network dimensionality. 0D + 2D = dimers of acid molecules and 2D stacks of paracetamol. | ||||||||
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 Paracetamol:anthranilic acid :1 Paracetamol:anthranilic acid | ||||||||
| CSP rank 1 | P21/c | 10.63 | 9.54 | 17.15 | 56.3 | −5593.30 | 1.324 | 3D |
| CSP rank 2 | P21/c | 13.30 | 6.24 | 17.78 | 77.6 | (+1.10) | 1.331 | 3D |
| CSP rank 3 | P21/c | 9.22 | 11.50 | 16.81 | 124.6 | (+1.51) | 1.304 | 3D |
| CSP rank 4 | P | 4.80 | 10.47 | 14.49 | 102.0 | (+1.58) | 1.345 | 1D |
| 90.6 | ||||||||
| 86.4 | ||||||||
| CSP rank 5 | P21/c | 4.78 | 27.82 | 11.42 | 109.8 | (+1.71) | 1.339 | 1D |
| CSP rank 6 | P | 4.80 | 8.02 | 19.60 | 80.5 | (+1.73) | 1.349 | 1D |
| 89.6 | ||||||||
| 107.1 | ||||||||
| CSP rank 7 | P21/c | 11.78 | 8.86 | 13.72 | 103.0 | (+1.73) | 1.372 | 2D |
| CSP rank 8 | P21/c | 4.82 | 10.37 | 27.93 | 91.2 | (+1.95) | 1.372 | 1D |
| CSP rank 9 | P21/c | 15.70 | 4.88 | 22.42 | 54.7 | (+1.95) | 1.367 | 1D |
| CSP rank 10 | P21/c | 14.53 | 8.48 | 11.56 | 80.1 | (+1.99) | 1.364 | 0D + 2D |
| 1:1 Paracetamol:benzoic acid | ||||||||
| CSP rank 1 | P21/c | 8.57 | 9.46 | 17.01 | 101.8 | −5301.82 | 1.344 | 2D |
| CSP rank 2 (3) | P21/c | 14.26 | 8.35 | 11.75 | 79.1 | (+0.21) | 1.321 | 0D + 2D |
| CSP rank 3 (2) | P21/c | 14.33 | 8.38 | 11.73 | 79.0 | (+0.22) | 1.312 | 0D + 2D |
| CSP rank 4 | P212121 | 4.49 | 11.33 | 26.64 | 90.0 | (+0.23) | 1.338 | 3D |
| CSP rank 5 | Pbca | 14.09 | 13.05 | 15.23 | 90.0 | (+0.23) | 1.297 | 0D + 2D |
| CSP rank 6 (9) | Pbca | 7.41 | 13.40 | 27.76 | 90.0 | (+0.35) | 1.317 | 0D + 2D |
| CSP rank 7 | P21/c | 5.68 | 11.73 | 20.46 | 100.1 | (+0.35) | 1.353 | 2D |
| CSP rank 8 | Pbca | 13.91 | 13.16 | 15.13 | 90.0 | (+0.37) | 1.311 | 0D + 2D |
| CSP rank 9 (6) | Pbca | 7.42 | 13.41 | 27.83 | 90.0 | (+0.42) | 1.311 | 0D + 2D |
| CSP rank 10 | P21/c | 14.56 | 8.49 | 11.53 | 75.8 | (+0.54) | 1.313 | 0D + 2D |
Fig. 4a shows a plot of relative lattice energies versus densities of the DFT-D predicted structures of anthranilic acid as well as the DFT-D optimised form I. The rank 1 and 2 predictions correspond to the experimental forms II and III respectively. The DFT-D optimised form I structure is 0.74 kcal mol−1 more stable than the rank 1 prediction. The thermodynamic relationships between the three experimental polymorphs are not clear in the literature. Upon heating, form I transforms into a new phase which is probably form II or III prior to melting.30 This observation implies that form I might be the most stable form at low temperature. Fig. 4b shows a similar plot of the predicted benzoic acid structures. Note that the acidic proton of the only polymorph reported in the CSD is disordered over two different sites with occupancies 0.87:0.13 at 20 K (CSD code: BENZAC07). The rank 1 and 2 predictions correspond to this disordered structure with the proton located in either site, and are isostructural to each other. Interestingly, eight out of ten low-energy predicted structures form four different isostructural pairs. A similar result was obtained in the CSP study of 5-chloroaspirin.31 For anthranilic acid, however, no isostructural pair is observed among the predicted structures.
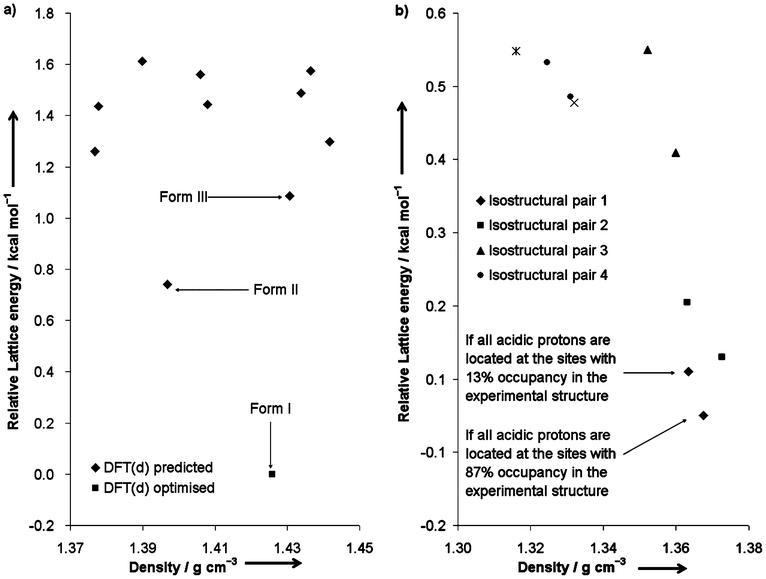 | ||
| Fig. 4 Plots of relative lattice energies versus densities of a) the DFT-D predicted structures of anthranilic acid and the DFT-D optimised form I, and b) the DFT-D predicted structures of benzoic acid. | ||
The lattice energies of the co-crystals are expressed in kilocalories per mole of formula unit. Their stabilities are calculated relative to the lowest-energy structures of the component molecules determined by the DFT-D method. Fig. 5a shows that the most stable predicted 1:1 paracetamol:anthranilic acid co-crystal is 1.68 kcal mol−1 less stable than the DFT-D optimised structures of paracetamol form I and anthranilic acid form I. This co-crystal structure will still be 0.94 kcal mol−1 less stable than the component molecules if the comparison is made without considering form I of anthranilic acid, which could not have been predicted in the CSP of that molecule. Fig. 5b shows that the most stable predicted 1:1 paracetamol:benzoic acid co-crystal is 1.71 kcal mol−1 less stable than paracetamol form I and the more stable form of the disordered benzoic acid structure. Hence, these results show that there exists a thermodynamic reason why paracetamol does not form co-crystals with these two acid molecules. If formed, the co-crystals are likely to be very unstable at low temperature. Note that, in seven out of ten predicted paracetamol:benzoic acid co-crystal structures, isolated dimers of benzoic acid molecules are embedded between two-dimensional hydrogen-bonded stacks of paracetamol. However, isolated anthranilic acid dimers are only observed in one of the predicted paracetamol:anthranilic acid co-crystal structures. The phenomenon may be attributed to the absence of an amino group in benzoic acid and the stoichiometric ratio of the component molecules in the predicted co-crystals.
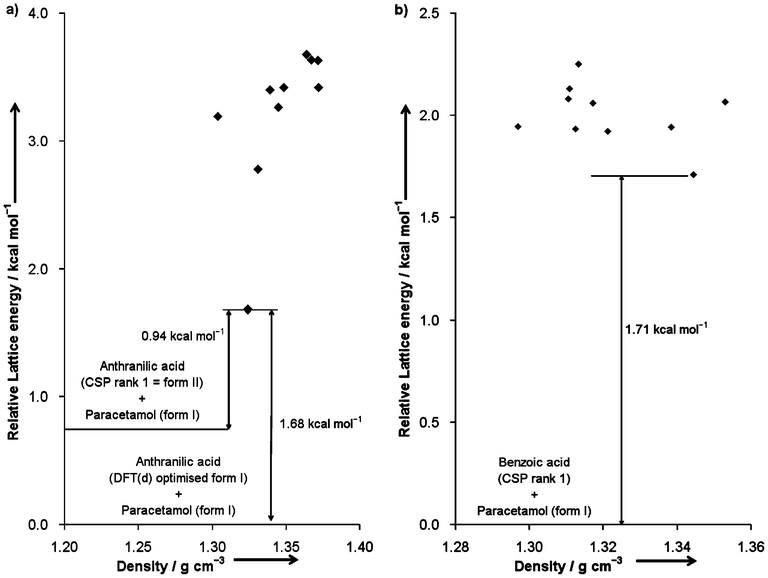 | ||
| Fig. 5 Plots of relative lattice energies versus densities of a) the DFT-D predicted 1:1 paracetamol:anthranilic acid co-crystals, and b) the DFT-D predicted 1:1 paracetamol:benzoic acid co-crystals. | ||
There is a chance that stoichiometries other than the 1:1 ratios studied here may produce more stable co-crystals,6 although they have not been observed experimentally for these compounds. This study once again highlights the challenge in predicting multi-component molecular crystals. The addition of an independent molecule in the structure search has led to a sharp increase in the number of structures to be considered, if the same high level of confidence for locating the DFT-D global minimum is required (Table 2). This problem is expected to be more profound in predicting co-crystals with different stoichiometric ratios. The CSP results also provide useful insights into the effects of functional-group substitution to a given molecule on the resulting hydrogen-bonding patterns and the crystal packings – an area of continuing interest in the crystal engineering community.
Conclusions
The thermodynamic stability of 102 co-crystals and salts of nicotinamide, isonicotinamide and picolinamide has been investigated by means of lattice energy calculations using the DFT-D method. In 99 (over 97%) of the 102 cases the calculated stability is in accord with the fact that the compound can be crystallised. This is the first study that provides conclusive evidence that the existence of salts and co-crystals and their stability relative to their co-formers can be predicted reliably.The correct prediction of a negative co-crystallisation result is the second essential component of an ab initio screening method for co-crystal formation. Therefore, this study investigated whether current state-of-the-art crystal structure prediction (CSP) methods can be used to predict that a certain co-crystal cannot be obtained by studying two co-crystals of paracetamol with anthranilic acid and benzoic acid, for which no co-crystals have been obtained experimentally despite considerable efforts. In agreement with experiment, the two co-crystals are predicted to be significantly less stable than their pure components.
The results presented here strengthen our previous findings10–13 that it is of paramount importance in computational studies to get the thermodynamics of crystallisation correct before invoking any kinetic arguments. This study provides confidence that, even when the crystal structures of co-crystals or their co-formers are not known, CSP using accurate lattice energy calculations is able to predict reliably the existence and stability of a co-crystal or salt relative to its single component crystals. Therefore, advanced CSP methodology has a strong potential to complement experimental co-crystal screening technologies.
Acknowledgements
We thank Dr. Ian Scowen for useful discussions on the celecoxib:nicotinamide co-crystal structure.References
- S. Karki, T. Friščić, L. Fábián, P. R. Laity, G. M. Day and W. Jones, Adv. Mater., 2009, 21, 3905–3909 CrossRef CAS.
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- S. L. Childs and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 4208–4211 CAS.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CAS.
- A. J. Cruz-Cabeza, G. M. Day and W. Jones, Chem.–Eur. J., 2008, 14, 8830–8836 CrossRef CAS.
- N. Issa, P. G. Karamertzanis, G. W. A. Welch and S. L. Price, Cryst. Growth Des., 2009, 9, 442–453 CAS.
- P. G. Karamertzanis, A. V. Kazantsev, N. Issa, G. W. A. Welch, C. S. Adjiman, C. C. Pantelides and S. L. Price, J. Chem. Theory Comput., 2009, 5, 1432–1448 CrossRef CAS.
- M. A. Neumann and M.-A. Perrin, J. Phys. Chem. B, 2005, 109, 15531–15541 CrossRef CAS.
- M. A. Neumann, F. J. J. Leusen and J. Kendrick, Angew. Chem., Int. Ed., 2008, 47, 2427–2430 CrossRef CAS.
- A. Asmadi, M. A. Neumann, J. Kendrick, P. Girard, M.-A. Perrin and F. J. J. Leusen, J. Phys. Chem. B, 2009, 113, 16303–16313 CrossRef CAS.
- J. Kendrick, F. J. J. Leusen, M. A. Neumann and J. van de Streek, Chem.–Eur. J., 2011, 17, 10736–10744 CrossRef CAS.
- H. C. S. Chan, J. Kendrick and F. J. J. Leusen, Angew. Chem., Int. Ed., 2011, 50, 2979–2981 CrossRef CAS.
- J. van de Streek and M. A. Neumann, CrystEngComm, 2011, 13, 7135–7142 RSC.
- G. M. Day, T. G. Cooper, A. J. Cruz-Cabeza, K. E. Hejczyk, H. L. Ammon, S. X. M. Boerrigter, J. S. Tan, R. G. Della Valle, E. Venuti, J. Jose, S. R. Gadre, G. R. Desiraju, T. S. Thakur, B. P. van Eijck, J. C. Facelli, V. E. Bazterra, M. B. Ferraro, D. W. M. Hofmann, M. A. Neumann, F. J. J. Leusen, J. Kendrick, S. L. Price, A. J. Misquitta, P. G. Karamertzanis, G. W. A. Welch, H. A. Scheraga, Y. A. Arnautova, M. U. Schmidt, J. van de Streek, A. K. Wolf and B. Schweizer, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 107–125 Search PubMed.
- D. A. Bardwell, C. S. Adjiman, Y. A. Arnautova, E. Bartashevich, S. X. M. Boerrigter, D. E. Braun, A. J. Cruz-Cabeza, G. M. Day, R. G. Della Valle, G. R. Desiraju, B. P. van Eijck, J. C. Facelli, M. B. Ferraro, D. Grillo, M. Habgood, D. W. M. Hofmann, F. Hofmann, K. V. J. Jose, P. G. Karamertzanis, A. V. Kazantsev, J. Kendrick, L. N. Kuleshova, F. J. J. Leusen, A. V. Maleev, A. J. Misquitta, S. Mohamed, R. J. Needs, M. A. Neumann, D. Nikylov, A. M. Orendt, R. Pal, C. C. Pantelides, C. J. Pickard, L. S. Price, S. L. Price, H. A. Scheraga, J. van de Streek, T. S. Thakur, S. Tiwari, E. Venuti and I. K. Zhitkov, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 535–551 Search PubMed.
- A. Belsky, M. Hellenbrandt, V. L. Karen and P. Luksch, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 364–369 CrossRef.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425–14432 CrossRef.
- P. Vishweshwar, A. Nangia and V. M. Lynch, Cryst. Growth Des., 2003, 3, 783–790 CAS.
- GRACE software from Avant-garde Materials Simulation, www.avmatsim.eu.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- D. J. Berry, C. C. Seaton, W. Clegg, R. W. Harrington, S. J. Coles, P. N. Horton, M. B. Hursthouse, R. Storey, W. Jones, T. Friščić and N. Blagden, Cryst. Growth Des., 2008, 8, 1697–1712 CAS.
- J. F. Remenar, M. L. Peterson, P. W. Stephens, Z. Zhang, Y. Zimenkov and M. B. Hickey, Mol. Pharmaceutics, 2007, 4, 386–400 CrossRef CAS.
- M. Habgood, M. A. Deij, J. Mazurek, S. L. Price and J. H. ter Horst, Cryst. Growth Des., 2010, 10, 903–912 CAS.
- M. A. Neumann and M.-A. Perrin, CrystEngComm, 2009, 11, 2475–2479 RSC.
- M. A. Neumann, J. Phys. Chem. B, 2008, 112, 9810–9829 CrossRef CAS.
- J. A. Chisholm and S. Motherwell, J. Appl. Crystallogr., 2005, 38, 228–231 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- W. H. Ojala and M. C. Etter, J. Am. Chem. Soc., 1992, 114, 10288–10293 CrossRef CAS.
- R. Montis, M. B. Hursthouse, H. C. S. Chan, J. Kendrick and F. J. J. Leusen, CrystEngComm, 2012, 14, 1672–1680 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of DFT-D methodology, relative stabilities of co-former polymorphs, calculated stabilities of co-crystals and salts, alternative celecoxib:nicotinamide co-crystal structures, CIF files with DFT-D optimised crystal structures of co-formers, co-crystals, salts, predicted structures and the proposed VIGDAR crystal structure. See DOI: 10.1039/c3ce40107c |
| This journal is © The Royal Society of Chemistry 2013 |