 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programming the assembly of carboxylic acid-functionalised hybrid polyoxometalates†
Marie
Hutin
,
Carine
Yvon
,
Jun
Yan
,
Andrew
Macdonell
,
De-Liang
Long
and
Leroy
Cronin
*
School of Chemistry, WestCHEM, Joseph Black Building, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Lee.Cronin@glasgow.ac.uk
First published on 21st January 2013
Abstract
We report here the straightforward synthesis and characterisation of a series Anderson-type hybrid polyoxometalates in high yield, functionalised with carboxylic acid following the reaction of anhydride precursors with the starting hybrid cluster ([n-N(C4H9)4]3[MnMo6O18((OCH2)3CNH2)2]). Seven new structures have been obtained, five of which have acid-terminated ligands. Six of these structures have been isolated with a yield higher than 80% with high purity. This reaction is limited by the bulkiness of the anhydride used; this effect can be employed to selectively synthesise one isomer out of three other possibilities. The acid groups and aromatic platforms attached to the clusters can act as building tools to bridge several length scales and engineer molecular packing within the crystal structure. The presence of acids should also change the hydrophilicity of the clusters, and therefore the way they interact with hydrophilic surfaces. We also show a potential relationship between the acid group interaction in the packing diagram and the cluster's tendency to interact with a hydrophilic surface. In addition to reporting a derived synthetic path to new acid-terminated Mn-Anderson-type hybrids, we describe here a new way to program self-assembly motifs of these compounds in the crystal structure and at interfaces.
Introduction
The self-assembly of Mo, W, V or Nb oxides leads to the formation of nanoclusters called polyoxometalates (POMs).1–3 They have attracted an increasing interest in the last couple of decades within the wide scientific community due to their potential applications in fields as diverse as medicine (anti-cancer, anti-viral agents or in neurodegenerative diseases),4–6 and materials for their electronic and magnetic properties.6–10 Their structural characteristics are as versatile, as are their electronic properties, but their full potential can only be reached if their systematic derivatisation and self-assembly is better understood, allowing the precise design and fine tuning of these sought-after features. Combining their diverse electronic properties with those of more established and well-understood organic moieties via “covalent”, non-dissociable bonds in solution leads to a specific class of organic–inorganic polyoxometalate-based hybrids.11–18 Despite an increasing interest in this field, their synthesis and diversification remain challenging; for instance, it is difficult to “program” in specific structural and physical properties. Although the potential of combining the properties of inorganic and organic subcomponents in a synergistic fashion is attractive, they require a different synthetic approach unique to this class of molecules.19,20Since 2003, where the first Mn-Anderson hybrid was reported,21 great progress has been made in the functionalisation of the amine-terminated hybrids.16,22–26 Two approaches can be used: pre-functionalisation which involves the reaction of the pre-synthesised tris-alkoxy chelating ligand with the inorganic precursor [Mo8O26]TBA4 (TBA = tetrabutylammonium); and the post-functionalisation which takes advantage of the reactivity of a specific group on the organic moiety of the hybrid cluster. As an example, nitro groups have been included in hybrid molecules; while they are easily reduced in organic chemistry to form amines, this becomes more challenging in the presence of highly oxidised clusters. Therefore amine-terminated hybrids are synthesised using a pre-functionalisation approach, which can be reacted afterwards to form amides or imines.
It is well-known that amines and hydroxyl groups react with anhydrides to form amides or esters, respectively. In 2011 Wei et al.27 reported the esterification of a series of hybrid polyoxovanadates via the reaction of anhydrides with a free alkoxy group on the hybrid in a post-functionalisation approach. However the reaction of anhydrides with the amine of the TRIS Mn-Anderson has not yet been reported. The use of cyclic anhydrides, instead of linear ones, should lead to the formation of acid-terminated hybrid Mn-Anderson, allowing us to develop a new route to programming the structures and properties of these clusters both in the solid state and at interfaces.16,28–33 Here we show that such hybrids are straightforwardly obtained in high yields (typically more than 80%) with high purity (based on elemental analyses) using a wide range of commercially available anhydride precursors, both cyclic (e.g. leading to acid-functionalised hybrids) and linear (see Scheme 1).
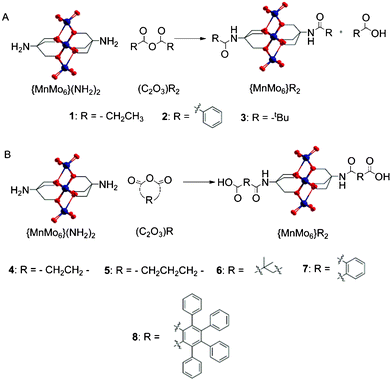 | ||
| Scheme 1 A. Linear anhydride (C2O3)R2 reacting with {MnMo6}(NH2)2 and B. Cyclic anhydride (C2O3)R reacting with {MnMo6}(NH2)2. Colour scheme: O (red), Mo (dark blue) and Mn (orange). | ||
In addition to bridging the molecular and macromolecular length scale with complementary motifs,34–36 these terminal acid groups should also change the hydrophilicity of the hybrid structures and therefore increase their interaction with hydrophilic surfaces, which should be seen by AFM.25,37–40 Gaining a better understanding of the surface interaction of these paramagnetic hybrids would be of particular relevance for the development of magnetically interesting hybrid-based thin films.33,41–43
Results and discussion
Synthesis
The reaction of the Mn-Anderson cluster [n-N(C4H9)4]3 [MnMo6O18((OCH2)3CNH2)2] in DMF (DMF = dimethylformamide) or MeCN (MeCN = acetonitrile) led to i) non-acid terminated compounds {MnMo6}R2 with the linear anhydrides (C2O3)R2 where {MnMo6} = TBA3[MnMo6O24((CH2)3CNHCO–)2] and R = 1 (–CH2CH3), 2 (–C6H5); and ii) led to acid-terminated compounds {MnMo6}R2 with the cyclic anhydrides (C2O3)R where {MnMo6} = TBA3[MnMo6O24((CH2)3CNHCO–)2(COOH)2] and R = 4 (–(CH2)2–), 5 (–(CH2)3–), 6 (–(C(CH3)2CH2)–), 7 (–(C6H4)–) and 8 (–(C6(C6H5)4)–) (see Scheme 1). Compounds were isolated in high yield following crystallisation with Et2O (Et2O = diethyl ether) vapour diffusion and subsequently recrystallised from MeCN or DMF for analytical purposes. All these compounds exhibited a high solubility in MeCN, DMF and DMSO. The compounds incorporating aromatic rings were less soluble in MeCN. None of these were soluble in water. The structure of these clusters and their arrangement in the packing diagrams have been analysed by crystallography.In Table 1 different reaction conditions are summarised for the syntheses of hybrids {MnMo6}R2 with R = 1–7. All these reactions have been performed with 50 (±1.5) mg of the hybrid precursor {MnMo6}(NH2)2 in 1 mL of solvent, DMF or MeCN, at 50 °C overnight. The products are then crystallised by Et2O vapour diffusion for 24 h or until the mother liquors are colourless (indicating that all the hybrid compound has crystallised), then the crystals are filtered, washed with Et2O and dried overnight under vacuum. The conversion ratios, in %, have been determined by integrating the NMR peaks of the ligand and comparing them with the NMR integration for the peaks of the TBA cations which are related to the total amount of hybrid clusters in solution, reacted or unreacted.
| (C2O3)R2 | (C2O3)R | ||||||
|---|---|---|---|---|---|---|---|
| R= | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| a Note. n. s. means non soluble. | |||||||
| 10 eq. MeCN | 100 (93.7%) | 100 (82.4%) | 9 | 94 | 94 | n. s. | 72 |
| 10 eq. DMF | 10.5 | 92 | 98.5 | 100 (86.6%) | 90 | ||
| 20 eq. DMF | 17.5 | 100 (96.4%) | 100 (95.3%) | 100 (79.5%) | |||
Linear and non-bulky anhydrides led in high yields in MeCN to the corresponding hybrids (see Table 1). When a bulkier anhydride is used such as the tert-butyl anhydride (C2O3)32 in the same reaction conditions, the total amount of starting material {MnMo6}(NH2)2 reacting is very low, about 9%, based on NMR spectra integrations. This rate can be increased by increasing the ratio of anhydride in DMF (see Table 1), although {MnMo6}32 could not be isolated and purified. Despite leading generally to products in high yields using a simple protocol, this reaction reaches its limitation with the bulkiness of the organic reactant used.
The main benefit of this reaction is the possibility to straightforwardly obtain the Anderson-type cluster functionalised with acid group, which should open the way to new Mn-Anderson hybrid using the carboxylic acid reactivity. This can be achieved similarly than previously described using a closed, cyclic anhydride such as the succinic, the glutaric, the dimethylsuccinic, and the phthalic anhydrides. These led in high yields and high purities to the corresponding acid terminated Mn-Anderson hybrids {MnMo6}42, {MnMo6}52, {MnMo6}62 and {MnMo6}72 respectively. Similarly to what has been observed for the linear anhydrides, the bulkiness of the anhydride used is a critical factor. Indeed, only a couple of crystals have been obtained from the reaction of (C2O3)8. In addition the obtention of {MnMo6}82 is scarcely reproducible with MS (MS = mass spectrometry) studies mainly showing the presence of the mono-substituted hybrid in the reaction mixture. Unfortunately we have not been able to isolate the corresponding asymmetric cluster.
Interestingly, what could appear as a limiting factor can be used to enhance the formation of a single regioisomer.
For instance, when the dimethylsuccinic anhydride (C2O3)6 is reacted with the hybrid precursor, three stereoisomers of {MnMo6}62 can be obtained (see Scheme 2), which should lead statistically to 25% of (a), 25% of (b) and 50% of (c). These three isomers cannot be distinguished by proton NMR in the normal range of frequencies (e.g. 20 to −5 ppm), but the peak corresponding to the –CH2 groups from the chelating part of the ligand found at around 60 ppm due to the influence of the paramagnetic manganese, shows the presence of the different clusters.
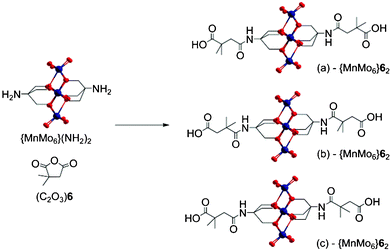 | ||
| Scheme 2 The three possible isomers of {MnMo6}62 obtained from (C2O3)6. Colour scheme: O (red), Mo (dark blue) and Mn (orange). | ||
When 10 equivalents of the anhydride are reacted in MeCN with {MnMo6}(NH2)2, three broad peaks are present in this area, one of which corresponding to the starting hybrid material. This was expected since the integration of the peak in the normal range of frequencies against those of the counter-cation showed that 72% of starting hybrid material had reacted under these conditions (Table 1). In addition, the 2 other peaks have nearly the same intensity, consistent with a close-to-the-statistics composition mixture. When the same reaction is performed in DMF, 90% of starting material was converted (Table 1), but no peak corresponding to the chelating CH2 of the starting material can be seen (Fig. 1), probably because it is too broad to be distinguished from the baseline. It is worth noting that one of the two remaining peaks is much more intense, showing that the solvent tends to favour one isomer. Finally, when 20 equivalents of the anhydride were reacted with {MnMo6}(NH2)2 in DMF, all the starting material was consumed and only one broad peak was seen at around 64 ppm, suggesting that one isomer is strongly favoured.
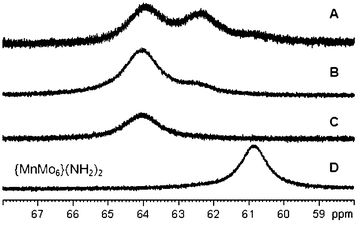 | ||
| Fig. 1 1H NMR spectra in d6-DMSO of {MnMo6}62 synthesised in different conditions. (C2O3)6 is reacted with {MnMo6}(NH2)2 following the conditions described in Table 1: A) 10 equivalent in MeCN, B) 10 equivalents in DMF, C) 20 equivalents in DMF and D) NMR of the hybrid precursor {MnMo6}(NH2)2 in d6-DMSO. | ||
Crystal structures
All the crystal structures were obtained either from MeCN–Et2O or DMF–Et2O. A summary of all the crystal structure parameters is provided in Table 2. Compound {MnMo6}12 has been isolated in 82.5% yield as orange crystals, and crystallises in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with Z = 2. The central Mn3+cations are chelated by the two tris-alkoxy moieties, featuring Mn–O bond lengths in the range 1.9139(16) to 2.0190(15) Å and MnO–C bond lengths in the range 1.426(2) to 1.437(2) Å. Unsurprisingly the hybrid units are wrapped around the TBA cations and solvent molecules via CH–O hydrogen bonds. This dense network of H-bonds shields the clusters and prevents them from interacting with each other. Compound {MnMo6}22 crystallises in the Pca21 space group with Z = 4. The bond lengths and angles measured from the clusters were within the range of values expected for Mn-Anderson clusters.
space group with Z = 2. The central Mn3+cations are chelated by the two tris-alkoxy moieties, featuring Mn–O bond lengths in the range 1.9139(16) to 2.0190(15) Å and MnO–C bond lengths in the range 1.426(2) to 1.437(2) Å. Unsurprisingly the hybrid units are wrapped around the TBA cations and solvent molecules via CH–O hydrogen bonds. This dense network of H-bonds shields the clusters and prevents them from interacting with each other. Compound {MnMo6}22 crystallises in the Pca21 space group with Z = 4. The bond lengths and angles measured from the clusters were within the range of values expected for Mn-Anderson clusters.
| R= | 1 | 2 | 4 | 5 | 6 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Solvent | MeCN–Et2O | DMF–Et2O | MeCN–Et2O | MeCN–Et2O | MeCN–Et2O | DMF–Et2O | DMF–Et2O | DMF–Et2O |
| Empirical formula | C72H147Mn Mo6N10O26 | C79H153Mn Mo6N8O29 | C68H138Mn Mo6N7O30 | C76H151Mn Mo6N10O30 | C72H146Mn Mo6N7O30 | C77H161Mn Mo6N8O33 | C78H146Mn Mo6N7O32 | C143H219Mn Mo6N10O37 |
| M/g mol-1 | 2199.6 | 2309.7 | 2164.4 | 2315.7 | 2220.5 | 2357.7 | 2324.6 | 3300.9 |
| Crystal system | Triclinic | Orthorhombic | Orthorhombic | Triclinic | Monoclinic | Monoclinic | Orthorhombic | Triclinic |
| Space group |
P |
Pca21 | Pnna |
P |
P21/n | P21/n | Pmna |
P |
| a/Å | 14.8991(11) | 18.0293(7) | 28.3641(5) | 14.9246(8) | 17.2400(6) | 17.8569(2) | 23.8498(6) | 23.5808(3) |
| b/Å | 16.8272(12) | 20.8575(8) | 23.7876(5) | 17.3492(10) | 27.2040(11) | 27.1013(3) | 14.5730(3) | 24.7645(3) |
| c/Å | 22.2685(17) | 26.7597(11) | 15.0216(3) | 20.5931(11) | 20.4735(8) | 21.2905(2) | 15.4775(4) | 29.9716(4) |
| α/° | 73.868(4) | 90 | 90 | 77.925(4) | 90 | 90 | 90 | 100.967(2) |
| β/° | 74.700(4) | 90 | 90 | 78.223(3) | 91.794(2) | 90.1110(10) | 90 | 103.918(2) |
| γ/° | 66.883(3) | 90 | 90 | 74.076(3) | 90 | 90 | 90 | 93.897(2) |
| V/Å3 | 4856.2(6) | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 062.9(7) 062.9(7) |
10135.3(3) |
4952.7(5) | 9597.3(6) | 10303.42(19) |
5379.4(2) | 16559.1(4) |
| Z | 2 | 4 | 4 | 2 | 4 | 4 | 2 | 4 |
| Density/Mg m−3 | 1.504 | 1.525 | 1.418 | 1.553 | 1.537 | 1.52 | 1.435 | 1.324 |
| Absorption coefficient | 0.946 | 0.919 | 0.908 | 0.935 | 0.961 | 0.902 | 0.862 | 0.585 |
| F (000) | 2268 | 4768 | 4448 | 2388 | 4576 | 4880 | 2392 | 6880 |
| Reflections collected | 69193 |
69503 |
78055 |
73984 |
74010 |
163257 |
43374 |
262887 |
| Unique | 19055 |
16825 |
9622 | 18773 |
18657 |
20213 |
5414 | 64939 |
| R(int) | 0.03 | 0.134 | 0.063 | 0.096 | 0.109 | 0.056 | 0.041 | 0.039 |
| Refined parameters | 1018 | 827 | 494 | 808 | 1055 | 1072 | 249 | 3214 |
| Goodness-of-fit on F2 | 1.046 | 1.056 | 1.065 | 1.095 | 1.004 | 1.037 | 1.086 | 0.986 |
| R 1 | 0.029 | 0.082 | 0.048 | 0.083 | 0.055 | 0.038 | 0.067 | 0.05 |
| wR2 (All) | 0.075 | 0.225 | 0.13 | 0.189 | 0.127 | 0.1 | 0.225 | 0.187 |
| Δρ max/min/eA3 | 0.76/−0.84 | 0.98/−1.02 | 0.78/−0.52 | 1.59/−1.16 | 0.83/−0.91 | 1.33/−0.66 | 1.11/−1.01 | 1.61/−0.77 |
The TBA cations and solvent molecules wrap the {MnMo6}22 clusters and the crystal packing diagrams reveal only a weak pi-stacking interaction between the benzene units of the ligand with a distance between the centroids of 4.09 Å, forming 1D chains (see Fig. 2). No hydrogen bonds between the aromatic cycle and the inorganic moieties are observed.
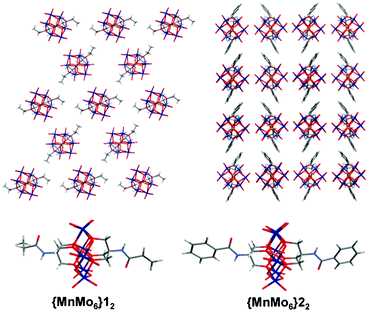 | ||
| Fig. 2 X-Ray structures of {MnMo6}12 (top and bottom left) and {MnMo6}22 (top and bottom right). The two packing diagrams are shown in the (yz) plan. Colour scheme: O (red), Mo (dark blue), Mn (orange), N (light blue), C (dark grey) and H (light grey). The TBA cations and the solvent molecules are removed from the packing diagrams for clarity. | ||
Compound {MnMo6}42 has been synthesised and isolated with a yield of 96.4% by crystallisation from a MeCN solution with Et2O vapour diffusion. The space group is Pnna with Z = 4, and the moieties –CH2CH2COOH are disordered (see ESI† and Fig. 3). Despite the presence of terminal acid groups, the clusters do not interact with each other and are wrapped by TBA cations.
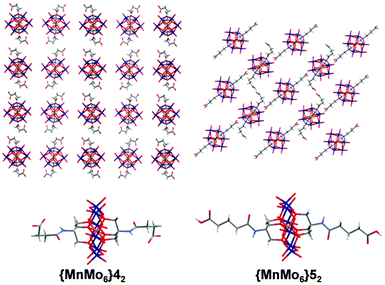 | ||
| Fig. 3 X-Ray Structures of {MnMo6}42 (top and bottom left, the structure is disordered) and {MnMo6}52 (top and bottom right). The packing diagram for {MnMo6}42 is shown in the (xy) plan and for {MnMo6}52 in the (yz) plan. The TBA cations and the solvent molecules are removed from the packing diagrams for clarity. Colour scheme: O (red), Mo (dark blue), Mn (orange), N (light blue), C (dark grey) and H (light grey). | ||
Compound {MnMo6}52 has been synthesised and isolated similarly than {MnMo6}42 in 95.3% yield. The space group is P with Z = 2. In contrast to {MnMo6}42, there are two different clusters in the unit. One features a torsion angle of 176(2)° between the first carbon after the amide group, the second, third carbon of the chain and the carbon of the acid. In the other hybrid, this angle is 76(2)°. These arrangements lead to a 3D network via two different types of hydrogen bond. The first one is an acid–acid interaction with a bond length of 2.629(19) Å. For the second one, these clusters are also linked by one oxygen from the inorganic moiety with an acid group from another type of cluster, the latter not being linked any further via their metallic part. The length of this OH–OMo bond is 3.476(18) Å.
Crystals of {MnMo6}62 suitable for X-ray analyses have been obtained from MeCN and DMF solutions with vapour diffusion of Et2O. In MeCN, only two types of crystals were observed, with needle-like and cubic shapes. Both correspond to the isomer (a) and crystallise with the same cell parameters. From MeCN and DMF the hybrid (a)-{MnMo6}62 crystallises in the P21/n space group with Z = 4. The change of solvent induces only slight changes in torsion angles and cell parameters. Two different torsion angles are observed between the carbon of the amide, the first and second carbons of the chain and the carbon of the acid: 60.2(6)° and 64.2(6)° from MeCN and 70.0(5)° and 66.6(4)° from DMF. The crystal packing (see Fig. 4) reveals that the two acid groups of each cluster are hydrogen-bond donors to oxygen atoms linked to a Mo forming a 3D H-bond network. The lengths of the acid–OMo bonds have been measured in the structure obtained from MeCN: 2.783(5) Å in the plan (yz), 3.927(5) Å in the plan (xy) and 3.377(5) Å in the plan (xz).
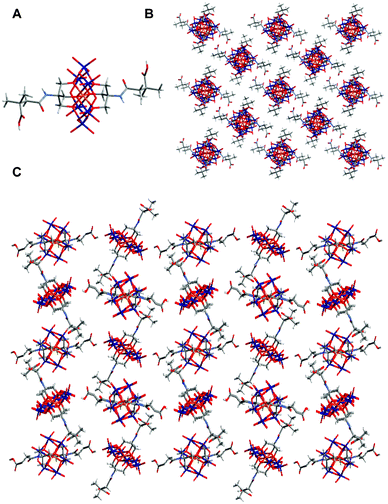 | ||
| Fig. 4 A) Crystal structure of (a)-{MnMo6}62; B) Packing diagram of (a)-{MnMo6}62 in the (xz) plan and C) evidence of the H-bonding network in the (yz) plan. Colour scheme: O (red), Mo (dark blue), Mn (orange), N (light blue), C (dark grey) and H (light grey). The TBA cations and the solvent molecules are removed from the packing diagrams for clarity. | ||
When the hybrid {MnMo6}(NH2)2 is reacted with the phthalic anhydride, the corresponding hybrid {MnMo6}72 is obtained in 79.5% yield. The compound crystallises from DMF by Et2O vapour diffusion (the solubility is too low in MeCN to grow crystals) in the space group Pmna with Z = 4 (Fig. 5). The aromatic cycles of the ligands are highly disordered, but it is clear from the packing diagram that the hybrid units are wrapped around the TBA cations and the solvent molecules and do not interact with each other (see ESI†).
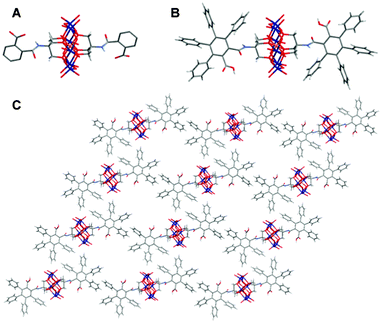 | ||
| Fig. 5 A) Crystal structure of {MnMo6}72; B) Crystal structure of {MnMo6}82 C) Packing diagram of {MnMo6}82. Colour scheme: O (red), Mo (dark blue), Mn (orange), N (light blue), C (dark grey) and H (light grey). The TBA cations and the solvent molecules are removed from the packing diagrams for clarity reasons. | ||
To obtain {MnMo6}82, {MnMo6}(NH2)2 was reacted with the tetraphenylphthalic anhydride (C2O3)8, for 10 days at 50 °C in DMF. From 100 mg of starting material, only a couple of crystals of this hybrid were obtained, and this reaction is hardly reproducible. The starting hybrid material crystallises in 24 hours, while it takes one or two weeks for {MnMo6}82, allowing it to be isolated from the starting material. Performing the reaction under inert atmosphere did not help to improve the yield and the reproducibility.
{MnMo6}82 crystallises in the space group P with Z = 2. Hydrogen bonds between the phenyl groups and the oxygens of the clusters maintain the structure in a 2D network. If the phenyl groups are numbered 1 to 4 from the amide group to the free acid group, the hydrogen in meta position on cycle 2 (Hmeta) form hydrogen bonds with an Mo–O and those in para position on this cycle (Hpara) form H bonds with Mo–O–Mo oxygens. Two clusters in the unit cell give two distinct sets of bond lengths. In one case, the hybrid is both donor and acceptor of H bond between Hmeta and Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, (3.542(3) Å and 3.568(3) Å), and between Hpara and Mo–O–Mo (3.406(3) Å and 3.450(3) Å). In the second case, the bond lengths are slightly larger featuring weaker interactions between Hmeta and MoO (3.897(3) Å and 3.911(2) Å), and between Hpara and Mo–O–Mo (3.426(3) Å and 3.470(3) Å). Surprisingly but similarly to what we have previously reported concerning a Mn-Anderson with aromatic ligands,26 no pi-stacking interactions are involved in the packing.
O, (3.542(3) Å and 3.568(3) Å), and between Hpara and Mo–O–Mo (3.406(3) Å and 3.450(3) Å). In the second case, the bond lengths are slightly larger featuring weaker interactions between Hmeta and MoO (3.897(3) Å and 3.911(2) Å), and between Hpara and Mo–O–Mo (3.426(3) Å and 3.470(3) Å). Surprisingly but similarly to what we have previously reported concerning a Mn-Anderson with aromatic ligands,26 no pi-stacking interactions are involved in the packing.
AFM studies
The hydrophilicity of the hybrids is changed by functionalising their organic moieties with carboxylic acid groups. It is therefore expected that: i) a given hybrid cluster will interact differently on glass surfaces with different hydrophilicities and ii) hybrids {MnMo6}12 and {MnMo6}22 will interact less than acid-functionalised hybrids {MnMo6}42, {MnMo6}52 and {MnMo6}62 on hydrophilic glass surfaces.Firstly, in order to determine how hydrophilic the glass surface needs to be for good interaction with the hybrid, 10 μL of a 1 mg mL−1 solution of {MnMo6}42 in MeCN has been drop cast on glass slides previously plunged in H2SO4:H2O2 solutions of composition 100:0, 7:1, 5:1 and 3:1 for 10 min. It is indeed known that increasing the proportion of sulfuric acid will increase the hydrophilicity of the surface.44 Unfortunately, the surfaces were too hydrophilic to measure the contact angles with an acceptable accuracy. The AFM height pictures reveal large isolated aggregates from the 3:1 solution, small densely packed aggregates from the 5:1 solution and flat flower-like features from the 7:1 solution, highlighting a higher surface interaction for the most hydrophilic surface (see ESI†). It is worth noting that the AFM pictures are not consistent over all the area of the surface, due to differences in local concentrations. However, the pictures have been taken as consistently as possible from one sample to another.
In order to assess the effect of the interaction between the glass surface and the hybrid molecule, solutions 1 mg mL−1 in MeCN of {MnMo6}12, {MnMo6}22, {MnMo6}42, {MnMo6}52 and {MnMo6}72 were drop cast (V = 10 μL) on glass slides previously cleaned in solutions H2SO4:H2O2 7:1, see in ESI† for more details on the procedure. After the drops were dried at ambient conditions, AFM height pictures were taken in the semi contact mode (see Fig. 6).
 | ||
| Fig. 6 Height AFM images taken in semi-contact mode: A) {MnMo6}12 (25 × 25 μm scan size), B) {MnMo6}22 (25 × 25 μm scan size), C) {MnMo6}42 (25 × 25 μm scan size), D) {MnMo6}42 (5 × 5 μm scan size), E) {MnMo6}52 (25 × 25 μm scan size) and F) {MnMo6}72 (5 × 5 μm scan size). | ||
Again, it is worth noting that the features observed depend on the local concentration.37 In the case of {MnMo6}12, the picture highlights a film with several holes, likely to be due to solvent evaporation. Except for these holes, the film is made of densely packed very small aggregates. In the case of {MnMo6}22, the picture shows a dense packing of very small dots of about 50 nm in height, and a diameter smaller than 1 μm. These two pictures seem to reveal poor surface interaction. Pictures of {MnMo6}42, on the other hand, reveal largely spread features of about 40 nm in height and several micrometers (typically 5 μm) in diameter. More interestingly, it features fractals or fibre-like sub-structures growing in ramification networks from the central nuclei. In addition, these large, flower-like structures do not overlap and are always separated by an empty space of a few nanometres (see profile extracts in ESI†). These motifs are rather flat with an average height of about 15–20 nm, but the “seeds” themselves are higher with a size of around 10 nm above the surface of the flower-like motifs. These reveal very good surface interaction, and as described earlier depend on the way the glass surface has been treated. This type of features has not been found in any of the other samples described here. This could result from the presence of the acid groups on the organic moieties of {MnMo6}42, allowing a good interaction with the hydroxyl groups of the glass surface, which cannot happen in case of {MnMo6}12 and {MnMo6}22.
Interestingly, going from {MnMo6}42 to {MnMo6}52, only one carbon is added to the organic moiety of the hybrid, but the pictures differ greatly. Indeed, the height pictures of {MnMo6}52 reveal a homogeneous film of small aggregates, where only a few holes due to solvent evaporation can be distinguished. Despite the size of the hole, it looks very similar to what was observed for {MnMo6}12, suggesting that despite having its organic moieties functionalised by an acid group, {MnMo6}52 does not exhibit a surface interaction equal to {MnMo6}42. Although this behaviour depends on the surface chemistry, it could be related to what was observed in the packing diagram: {MnMo6}42 crystallised without any interaction with other hybrid molecules and {MnMo6}52 exhibited a tight 3D hydrogen-bonding network. Because the acid group of {MnMo6}52 interact strongly with the inorganic part of the hybrid and with other acid groups, these are less likely to interact with the glass surface by hydrogen-bonding, whereas in the case of {MnMo6}42 these hydrogen bonds in solution must be inexistent or very weak, therefore showing a greater tendency to interact with the surface.
This hypothesis could be confirmed by what is observed on the AFM pictures of {MnMo6}72. Two different features are observed: flat and large structures highlighting fibre-like ramifications, much more regular in size that those of {MnMo6}42, and small and large aggregates of about 40 nm heights. In the packing diagram the clusters were shown as isolated structures not interacting with each other with free acid groups, explaining the presence of these flat motifs. However in solution, the acid – amide groups can partly condense and form phthalimides which can be noticed by MS experiments (see ESI†), therefore fewer acid groups are available to interact with the surface.
Conclusions
The reaction of the Mn-Anderson hybrid polyoxometalate substituted by an amine group with anhydride precursors leads to a new series of carboxylic acid functionalised clusters in high yields. We have shown that the steric hindrance around the carbonyl of the anhydride is a crucial limiting factor for this reaction, however this can be used to synthesise one regioisomer with great selectivity. In addition, we have shown that the acid groups can be used as recognition motifs to form hydrogen bonds in the crystal packing, provided that the chain bearing the acid is long enough. These new clusters can bind to hydrophilic glass surfaces, as long as these acid groups are not involved in stronger hydrogen-bonding interaction with other clusters in solution. Understanding how these electro- and photoactive molecules interact with the surface is of particular relevance in fields such as solar cells. Finally, because the carbonyl of the anhydride should exhibit different electrophilicity depending on the group attached to the anhydride, it should be possible to direct this reaction to yield asymmetric clusters by mixing two different anhydrides. This is currently under investigation and will be the subject of further work.Experimental
General remarks
[n-N(C4H9)4][α-Mo8O26] and [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CNH2}2] were synthesised according to literature methods.21,45Acid anhydrides were obtained from commercial sources and used without further purificationPreparation of {MnMo6}12
To a 14 mL vial is added 112.6 mg (1 eq.) of {MnMo6}(NH2)2, 77 μL (10 eq.) of propionic anhydride and 2 mL of MeCN. The reaction mixture is stirred at 50 °C overnight. Orange-yellow crystals are obtained after 24 h with Et2O vapour diffusion. The crystals are washed with Et2O and dried for 48 h under vacuum. Yield = 93.7% (111.8 mg). 1H NMR (d6-DMSO, 400 MHz, ppm): 0.94 (36H + 6H, –CH3 from TBA+ + –CH3 from ligand), 1.31 (24H, –CH2 from TBA+), 1.57 (24H, –CH2 from TBA+), 2.40 (4H, br, –CH2), 3.16 (24H, –CH2 from TBA+), 7.37 (2H, br, NH), 64.00 (–CH2, br). Elemental analysis: Calc. for C62H132MnMo6 O26N5 (1994.31 g mol−1): C, 37.34; H, 6.67; N, 3.51. Found: C, 37.40; H, 6.63; N, 3.57. ESI-MS (MeCN, negative mode): 1751.0 ([M-TBA]−, [2M-2TBA]2−).Preparation of {MnMo6}22
To a 14 mL vial is added 113.3 mg (1 eq.) of {MnMo6}(NH2)2, 137.6 mg (10 eq.) of benzoic anhydride and 2 mL of MeCN. The reaction mixture is stirred at 50 °C overnight. The precipitate is removed by filtration on a cotton plug in a Pasteur pipette. Orange crystals are obtained after 24 h with Et2O vapour diffusion. The crystals are washed with Et2O and dried for 48 h under vacuum. Yield = 82.4% (103.7 mg). 1H NMR (d6-DMSO, 400 MHz, ppm): 0.93 (36H, –CH3 from TBA+), 1.31 (24H, –CH2 from TBA+), 1.57 (24H, –CH2 from TBA+), 3.16 (24H, –CH2 from TBA+), 7.43 (6H, m), 7.6 (2H, br, s), 7.77 (4H, m), 64.30 (–CH2, br). Elemental analysis: Calc. for C70H132MnMo6O26N5 (2090.39 g mol−1), %: C 40.22, H 6.36, N 3.35. Found: C 40.26, H 6.41, N 3.40. ESI-MS (MeCN, negative mode): 1848.1 ([M-TBA]−, [2M-2TBA]2−).Preparation of {MnMo6}42
To a 14 mL vial is added 51.9 mg (1 eq.) of {MnMo6}(NH2)2, 55.4 mg (20 eq.) of succinic anhydride and 1 mL of DMF. The reaction mixture is stirred at 50 °C overnight. Orange-yellow crystals are obtained after 24 h with Et2O vapour diffusion. The crystals are washed with Et2O and dried for 48 h under vacuum. For elemental analyses they are afterwards recrystallised by Et2O vapour diffusion from MeCN. Yield = 96.4% (55.4 mg). 1H NMR (d6-DMSO, 400 MHz, ppm): 0.94 (36H, –CH3 from TBA+), 1.31 (24H, –CH2 from TBA+), 1.57 (24H, –CH2 from TBA+), 2.37 (4H, br, –CH2), 2.70 (4H, br, –CH2), 3.16 (24H, –CH2 from TBA+), 7.52 (2H, br, NH), 12.07 (2H, s, acid), 64.16 (–CH2, br). Elemental analysis: Calc. for C64H132MnMo6N5O30 (2082.33 g mol−1): C, 36.91; H, 6.39; N, 3.36. Found: C, 36.67; H, 6.55; N, 3.63. ESI-MS (MeCN, negative mode): 1840 ([M-TBA]−, [2M-2TBA]2−).Preparation of {MnMo6}52
To a 14 mL vial is added 105.4 mg (1 eq.) of {MnMo6}(NH2)2, 127.9 mg (20 eq.) of glutaric anhydride and 2 mL of DMF. The reaction mixture is stirred at 50 °C overnight. Orange crystals are obtained after 24 h with Et2O vapour diffusion. The crystals are washed with Et2O and dried for 48 h under vacuum. For elemental analyses they are afterwards recrystallised by Et2O vapour diffusion from MeCN. Yield = 95.3% (112.6 mg). 1H NMR (d6-DMSO, 400 MHz, ppm): 0.95 (36H, –CH3 from TBA+), 1.33 (24H, –CH2 from TBA+), 1.55 (24H, –CH2 from TBA+), 1.71 (4H, br, –CH2), 2.21 (4H, br, –CH2), 2.46 (4H, br, –CH2), 3.16 (24H, –CH2 from TBA+), 7.36 (2H, br, NH), 12.00 (2 H, s, acid), 64.53 (–CH2, br). Elemental analysis: Calc. for C66H136MnMo6N5O30 (2110.38 g mol−1): C, 37.56; H, 6.50; N, 3.32. Found: C, 37.41; H, 6.66; N, 3.87. ESI-MS (MeCN, negative mode): 1868.1 ([M-TBA]−, [2M-2TBA]2−).Preparation of {MnMo6}62
To a 14 mL vial is added 104.4 mg (1 eq.) of {MnMo6}(NH2)2, 144.8 mg (20 eq.) of dimethylsuccinic anhydride and 2 mL of DMF. The reaction mixture is stirred at 50 °C overnight. Orange crystals are obtained after 24 h with Et2O vapour diffusion. The crystals are washed with Et2O and dried for 48 h under vacuum. For elemental analyses they are afterwards recrystallised from MeCN by Et2O vapour diffusion. Yield = 86.6% (102.7 mg). 1H NMR (d6-DMSO, 400 MHz, ppm): 0.94 (36H, –CH3 from TBA+), 1.14 (12H, s, –CH3), 1.31 (24H, –CH2 from TBA+), 1.57 (24H, –CH2 from TBA+), 2.71 (4H, br, –CH2), 3.16 (24H, –CH2 from TBA+), 7.37 (2H, br, NH), 12.04 (2H, s, acid), 64.52 (–CH2, br). Elemental analysis: Calc. for C68H140MnMo6N5O30 (2138.43 g mol−1): C, 38.19; H, 6.60; N, 3.28. Found: C, 38.13; H, 6.68; N, 3.22. ESI-MS (MeCN, negative mode): 1896.1 ([M-TBA]−, [2M-2TBA]2−).Preparation of {MnMo6}72
To a 14 mL vial is added 57.2 mg (1 eq.) of {MnMo6}(NH2)2, 50.5 mg (10 eq.) of phthalic anhydride and 1 mL of DMF. The reaction mixture is stirred at 50 °C overnight. Orange crystalline needles are obtained after 24 h with Et2O vapour diffusion. The crystals are washed with Et2O and dried for 48 h under vacuum. Yield = 79.5% (52.6 mg). 1H NMR (d6-DMSO, 400 MHz, ppm): 0.93 (36H, –CH3 from TBA+), 1.31 (24H, –CH2 from TBA+), 1.57 (24H, –CH2 from TBA+), 3.16 (24H, –CH2 from TBA+), 7.32 (2H, br), 7.43 (2H, dd), 7.53 (2H, br), 7.74 (2H, d), 13.25 (2H, s, acid), 64.11 (–CH2, br). Elemental analysis: Calc. for C72H132MnMo6N5O30 (2178.41 g mol−1): C, 39.47; H, 6.08; N, 3.35. Found: C, 39.47; H, 6.18; N, 3.36. ESI-MS (MeCN, negative mode): 1936.0 ([M-TBA]−, [2M-2TBA]2−), 1676.7 ([M-H2O–TBA+H]−).X-Ray crystal structure determination
Suitable single crystals of compound 1–8 were attached to a thin glass fibber by using Fomblin YR-1800 oil and mounted on a goniometer head in a general position. All data were collected at 150 K on a Oxford Gemini A Ultra or a Bruker APEX II Quasar CCD diffractometers, with graphite monochromated X-radiation (Mo Kα, λ = 0.71073Å), running under the CrysAlisPro or Apex2 software. The corrections for the incident and diffracted beam absorption effects were applied using empirical or analytical methods.46,47 Structures were solved using Patterson or Direct methods with SHELXS-9748 or SIR-92 using WinGX routines. Structure refinement was accomplished by full matrix least-squares on F2via SHELXL-97.48 Most non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated using standard geometric criteria and refined on riding model. All data manipulation and presentation were performed using WinGX.49 Details of crystal data and the structure refinement are given in Table 2.Acknowledgements
The author thanks the EPSRC and the University of Glasgow for funding support, Jim McIver for technical support.References
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef.
- D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC.
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef CAS.
- J. T. Rhule, C. L. Hill and D. A. Judd, Chem. Rev., 1998, 98, 327 CrossRef CAS.
- J. Geng, M. Li, J. Ren, E. Wang and X. Qu, Angew. Chem., Int. Ed., 2011, 50, 4184 CrossRef CAS.
- H. E. Moll, W. Zhu, E. Oldfield, L. M. Rodriguez-Albelo, P. Mialane, J. Marrot, N. Vila, I. M. Mbomekallé, E. Rivière, C. Duboc and A. Dolbecq, Inorg. Chem., 2012, 51, 7921 CrossRef.
- E. Coronado and C. J. Gómez-Garcia, Chem. Rev., 1998, 98, 273 CrossRef CAS.
- J. M. Poblet, X. López and C. Bo, Chem. Soc. Rev., 2003, 32, 297 RSC.
- C. L. Hill, J. Mol. Catal. A: Chem., 2007, 262, 2 CrossRef CAS.
- N. Anwar, T. McCormac, J. D. Compain, P. Mialane, A. Dolbecq and F. Laffir, Electrochim. Acta, 2012, 59, 1 CrossRef CAS.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS.
- (a) P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77 CrossRef CAS; (b) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- A. Dolbecq, P. Mialane, L. Lisnard, J. Marrot and F. Sécheresse, Chem.–Eur. J., 2003, 9, 2914 CrossRef CAS.
- G. Rousseau, O. Oms, A. Dolbecq, J. Marrot and P. Mialane, Inorg. Chem., 2011, 50, 7376 CrossRef CAS.
- J. Rieger, T. Antoun, S.-H. Lee, M. Chenal, G. Pembouong, J. L. de la Haye, I. Azcarate, B. Hasenknopf and E. Lacôte, Chem.–Eur. J., 2012, 18, 3355 CrossRef CAS.
- Y. Yan, H. Wang, B. Li, G. Hou, Z. Yin, L. Wu and V. W. W. Yam, Angew. Chem., Int. Ed., 2010, 49, 9233 CrossRef CAS.
- C. Musumeci, M. H. Rosnes, F. Giannazzo, M. D. Symes, L. Cronin and B. Pignataro, ACS Nano, 2011, 5, 9992 CrossRef CAS.
- Y.-F. Song, D.-L. Long, S. E. Kelly and L. Cronin, Inorg. Chem., 2008, 47, 9137 CrossRef CAS.
- S. Thorimbert, B. Hasenknopf and E. Lacôte, Isr. J. Chem., 2011, 51, 275 CrossRef CAS.
- L. Zhu, Y. Zhu, X. Meng, J. Hao, Q. Li, Y. Wei and Y. Lin, Chem.–Eur. J., 2008, 14, 10923 CrossRef CAS.
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2406 CrossRef CAS.
- C. P. Pradeep, F.-Y. Li, C. Lydon, H. N. Miras, D.-L. Long, L. Xu and L. Cronin, Chem.–Eur. J., 2011, 17, 7472 CrossRef CAS.
- Y.-F. Song, D.-L. Long, C. Ritchie and L. Cronin, Chem. Rec., 2011, 11, 158 CrossRef CAS.
- E. F. Wilson, H. N. Miras, M. H. Rosnes and L. Cronin, Angew. Chem., Int. Ed., 2011, 50, 3720 CrossRef CAS.
- M. H. Rosnes, C. Musumeci, C. P. Pradeep, J. S. Mathieson, D.-L. Long, Y.-F. Song, B. Pignataro, R. Cogdell and L. Cronin, J. Am. Chem. Soc., 2010, 132, 15490 CrossRef CAS.
- Y.-F. Song, D.-L. Long and L. Cronin, CrystEngComm, 2010, 12, 109 RSC.
- P. Wu, Z. Xiao, J. Zhang, J. Hao, J. Chen, P. Yin and Y. Wei, Chem. Commun., 2011, 47, 5557 RSC.
- S. Zhang, J. Zhao, P. Ma, J. Niu and J. Wang, Chem.–Asian J., 2012, 7, 966 CrossRef CAS.
- K. Micoine, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Org. Lett., 2007, 9, 3981 CrossRef CAS.
- S. Bareyt, S. Piligkos, B. Hasenknopf, P. Gouzerh, E. Lacôte, S. Thorimbert and M. Malacria, J. Am. Chem. Soc., 2005, 127, 6788 CrossRef CAS.
- Z. He, H. Wang, Y. Wang, Y. Wu, H. Li, L. Bi and L. Wu, Soft Matter, 2012, 8, 3315 RSC.
- K. Nomiya, Y. Togashi, Y. Kasahara, S. Aoki, H. Seki, M. Noguchi and S. Yoshida, Inorg. Chem., 2011, 50, 9606 CrossRef CAS.
- Y. Yang, Y. Wang, H. Li, W. Li and L. Wu, Chem.–Eur. J., 2010, 16, 8062 CAS.
- P. Mialane, A. Dolbecq and F. Sécheresse, Chem. Commun., 2006, 3477 RSC.
- L. Lisnard, A. Dolbecq, P. Mialane, J. Marrot, E. Codjovi and F. Sécheresse, Dalton Trans., 2005, 3913 RSC.
- S.-T. Zheng, H. Zhang and G.-Y. Yang, Angew. Chem., Int. Ed., 2008, 47, 3909 CrossRef CAS.
- C. Musumeci, A. Luzio, C. P. Pradeep, H. N. Miras, M. H. Rosnes, Y.-F. Song, D.-L. Long, L. Cronin and B. Pignataro, J. Phys. Chem. C, 2011, 115, 4446 CAS.
- M. Jiang, X. D. Zhai and M. H. Liu, Langmuir, 2005, 21, 11128 CrossRef CAS.
- M. Jiang and M. Liu, J. Colloid Interface Sci., 2007, 316, 100 CrossRef CAS.
- M. Jiang, X. Zhai and M. Liu, J. Mater. Chem., 2007, 17, 193 RSC.
- P. J. Jensen and K. H. Bennemann, Surf. Sci. Rep., 2006, 61, 129 CrossRef CAS.
- H. E. Moll, A. Dolbecq, J. Marrot, G. Rousseau, M. Haouas, F. Taulelle, G. Rogez, W. Wernsdorfer, B. Keita and P. Mialane, Chem.–Eur. J., 2012, 18, 3845 CrossRef.
- M. Clemente-León, E. Coronado, C. L. Gómez-García, C. Mingotaud, S. Ravaine, G. Romualdo-Torres and P. Delhaès, Chem.–Eur. J., 2005, 11, 3979 CrossRef.
- J. J. Cras, C. A. Rowe-Taitt, D. A. Nivens and F. S. Ligler, Biosens. Bioelectron., 1999, 14, 683 CrossRef CAS.
- W. G. Klemperer, in Inorganic Syntheses, ed. A. P. Ginsberg, John Wiley & Sons Inc., 1990, vol. 27, p. 74 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887 CrossRef.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and ESI-MS spectra of compounds {MnMo6}R2R = 1, 2, 4, 5, 6, 7; crystallographic data for compounds {MnMo6}R2 with R = 1, 2, 4, 5, 6, 7, 8; packing diagrams for {MnMo6}R2 with R = 4 and 7, and with R = 6 from DMF–Et2O solution; AFM height pictures of {MnMo6}42 on glass slides with different hydrophilicities; profile extract for AFM pictures of {MnMo6}42 and {MnMo6}72. CCDC 909163–909170. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce26816k |
| This journal is © The Royal Society of Chemistry 2013 |