 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloidal systems for crystallization processes from liquid phase
Rafael
Muñoz-Espí
*a,
Yitzhak
Mastai
b,
Silvia
Gross
c and
Katharina
Landfester
a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: munoz@mpip-mainz.mpg.de
bDepartment of Chemistry and Institute of Nanotechnology and Advanced Materials, Bar-Ilan University, Ramat-Gan, 52900, Israel
cIstituto di Scienze e Tecnologie Molecolari, (ISTM-CNR), Dipartimento di Scienze Chimiche and INSTM, UdR Padova, Università degli Studi di Padova, Via Marzolo 1, 35131, Padua, Italy
First published on 29th January 2013
Abstract
Colloidal systems are involved in crystallization processes in many different ways: on one hand, colloids may be used as controlling or structure-directing agents, as nanoreactors and as scaffolds and templates for crystallization. On the other hand, it is very often desirable to obtain colloidal particles in a crystalline or nanocrystalline state. In this highlight, we cover the challenges and the recent advances on the following topics: (i) the use of polymer colloids as additives for crystallization due to their ability to control nucleation and growth and even to promote enantiomer resolution by enantioselective crystallization; (ii) state of the art in the preparation of inorganic crystalline colloids; and (iii) the application of colloidal systems (i.e., colloidal particles, droplets, micelles and vesicles) as supports, templates and nanoreactors for inorganic crystallization.
 Rafael Muñoz-Espí | Rafael Muñoz-Espí studied chemistry at the University of València. He received his doctoral degree from the Johannes Gutenberg University of Mainz in 2006, after working with Prof. Gerhard Wegner at the Max Planck Institute for Polymer Research. In 2007, he moved to the State University of New York at Stony Brook, where he worked for two years as a postdoctoral associate in the group of Prof. Benjamin Chu. Since 2009, he has been a project leader at the Max Planck Institute for Polymer Research in the department headed by Prof. Katharina Landfester. His research interests include mineralization processes, crystallization in colloidal systems and the study of the interaction of polymers with inorganic matter. |
 Yitzhak Mastai | Yitzhak Mastai obtained his BSc in physical chemistry from the Bar-Ilan University in 1989 and received his PhD in 1999 from the Weizmann Institute of Science, after working with Prof. Gary Hodes on nanomaterials synthesis. He then moved as a postdoctoral fellow to the Max Planck Institute of Colloids and Interfaces in Golm to work for 3 years with Prof. M. Antonietti and H. Cölfen on biomimetic chemistry and chiral polymers. In 2003, he joined the Chemistry Department of the Bar-Ilan University, where he is currently a Professor at the Institute of Nanotechnology. His current research is focused on the synthesis and analysis of chiral nanosurfaces, chiral self-assembled monolayers and polymeric chiral nanoparticles. |
 Silvia Gross | Silvia Gross received a degree in chemistry (110/110 cum laude) in 1996 and obtained her PhD in the same discipline in 2000 from the University of Padua. In 2001, she moved to Vienna for a 2-year post-doctoral stay, working on hybrid materials. In 2002, she was appointed a research scientist of the Institute of Molecular Science and Technology of the Italian National Research Council (CNR) and since 2010 she has been a senior researcher at the same institute. She spent different periods as a visiting scientist at the University of Stuttgart (2004, 2005) and Versailles (2007, 2008). Her scientific activity, focused on wet-chemistry routes for inorganic molecular and bulk nanosystems and on organic–inorganic hybrid materials, is documented by about 90 ISI-listed scientific publications. |
 Katharina Landfester | Katharina Landfester studied chemistry at the Technical University of Darmstadt. In 1995, she received her doctoral degree in physical chemistry from the Johannes Gutenberg University of Mainz after working with Prof. H.W. Spiess at the Max Planck Institute for Polymer Research. For a postdoctoral stay, she moved to the Lehigh University (Prof. M. El-Aasser), where she first came in contact with the miniemulsion technique. She returned to Germany in 1998, joining the group of Prof. M. Antonietti at the Max Planck Institute of Colloids and Interfaces in Golm. In 2002, she got her habilitation at the University of Potsdam. In 2003, she accepted a chair (C4) of macromolecular chemistry at the University of Ulm. Since 2008, she has been a director at the Max Planck Institute for Polymer Research. |
1. Introduction
According to IUPAC's Gold Book, in a colloidal system, “the molecules or polymolecular particles dispersed in a medium have at least in one direction a dimension of roughly between 1 nm and 1 μm” or, alternately, “discontinuities are found at distances of that order”.1 This size range implies that colloid science, a centenarian discipline, falls almost as a whole under the domain of the much younger and presumably “sexier” field of nanotechnology. In reality, colloids and thus nanomaterials have been known for centuries, even for millennia. The most recurrent example of ancient nanotechnology, used over and over again in written texts and academic presentations on the topic, is the Lycurgus Cup, a more than 1700 year-old piece manufactured by Roman craftsmen and exhibited at the British Museum in London. We will also allow ourselves to mention here once more this fascinating glass cup because it involves the two aspects of the focus of this highlight: colloidal systems and crystals. The Lycurgus Cup contains a colloidal crystalline gold–silver alloy, which explains why the opaque green color changes to glowing translucent red under illumination.2It is certainly true that nanocrystalline colloids find many technological applications in fields as diverse as optics, electronics and medicine,3–5 and we will also consider in the next pages the most recent advances in this area. However, we aim to emphasize here that the importance of colloidal systems in crystallization processes goes much beyond conventional nanocrystalline colloids. Colloidal systems can also act as controlling or structure-directing agents, nanoreactors and as scaffolds or templates for crystal formation.
We will limit the discussion to crystallization processes from the liquid phase, which already provide a broad enough scope, leaving aside physical or solid-state methods. Thus, we will also not refer directly to the so-called “colloidal crystals”, understood as 2D and 3D arrays of monodisperse noncrystalline colloids.
In the first part, we will focus on the use of colloids—mostly polymer colloids—as additives and controlling agents in crystallization processes of inorganic but also organic materials. How colloids can also be used to promote enantioselective crystallization will also be described. Afterwards, we will review the preparation of crystalline inorganic colloids by conventional wet-chemical methods. In the last part, the templating effect of colloidal systems will be discussed, not only the use of hard templates, but also the case of soft templating. Under “hard templates”, we will highlight an emerging topic of the last few years: the crystallization of inorganic nanoparticles on the surface of bigger colloidal particles, especially the case of covalently functionalized particles. These particles are used either as sacrificial templates or as a support to obtain hybrid materials. Finally, we will show how colloidal systems act as “soft templates”, confining spaces in which crystallization can take place. As an overview, the scenario of colloidal systems in and for crystallization processes, presented within this article, is schematically depicted in Fig. 1.
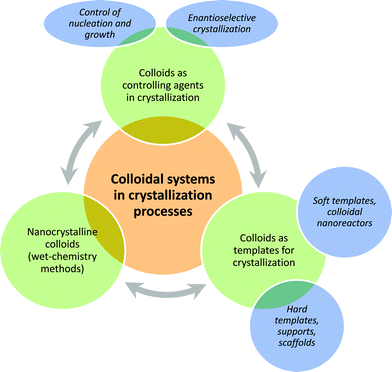 | ||
| Fig. 1 Scenario of the colloidal systems in crystallization processes. | ||
2. Colloidal systems as controlling agents for crystallization
It is well known that additives can affect the crystal formation in different ways: they can influence solubility–precipitation equilibria, nucleation, growth, Ostwald ripening, aggregation or can even cause stabilization of certain phases or polymorphs. Accordingly, additives are very often used in crystal engineering and have been the object of extensive studies.6,7 Nature provides very beautiful examples of how crystalline materials can be controlled by the presence of very diverse additives. Biomineralization processes can indeed be considered as additive-controlled processes of crystal formation.Cölfen and coworkers have contributed very prominently to the understanding of the effect of biogenic molecules and polymers in biomineralization and bioinspired crystallization.7–10 One of the most important ideas arisen in the last few years is that, very often in nature and in many synthetic processes, crystal formation takes place through a highly parallel “oriented attachment” of nanoparticles, forming so-called “mesocrystals”.11–13 Cölfen and Antonietti have named the concept as “non-classical crystallization”,11,13 in contraposition to the classical crystallization. In the “classical theory”, ions or single molecules form clusters until they reach a nucleus of critical size, being governed by solubility products and diffusion processes. The “non-classical crystallization” theory—which had been around in a latent state for years, but had never been formulated as such—is very important in terms of colloidal science because it states that biomineralization and many processes of additive-controlled crystallization occur through the colloidal pathways of oriented attachment of existing nanoparticles. These superstructures of nanoparticles can incorporate foreign additives with the ability to control the growth. Intriguing structures in nature with very complex morphologies but behaving like single crystals could be now explained in terms of “mesocrystals”.
2.1. Polymer colloids as additives for inorganic crystallization
Since the rate of the growth is frequently controlled at the interface, the adsorption of foreign substances at growing sites can significantly affect the crystal growth. Impurities may specifically adsorb onto certain growing faces, enhancing or retarding growth rates along certain directions and determining the final morphology of the crystals. These “classical” statements are also valid in the formation of mesocrystalline structures. The attachment of foreign substances can take place at kink sites, at a step or at a ledge (face) between steps, as depicted in Fig. 2. Although the original sketch shown in this figure, by Davey and Mullin,14 was proposed for ionic and small molecular additives, it is perfectly extendable to bigger and more complex species, such as colloidal additives.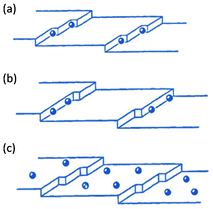 | ||
| Fig. 2 Adsorption of foreign substances on a crystal surface: (a) at a kink site, (b) at a step and (c) on an edge. (Reprinted from ref. 14, Copyright 1976, with permission from Elsevier.) | ||
Small molecules and polymers, including biomacromolecules, are the most common additives for crystallization, but different forms of colloidal systems can also be used to control nucleation and growth. Initiated by Wegner and coworkers in the early 2000s15 and followed up in the subsequent years, surface-functionalized polystyrene latex nanoparticles have been used as additives in the crystallization of zinc oxide,16,17 calcium carbonate18–20 and calcium oxalate.21 As latex particles can be easily visualized by electron microscopy, one of the initial motivations to use them as controlling agents for crystallization was to prove the incorporation of polymer additives into the crystal structure. The results confirmed the hypothesis (see Fig. 3) and the holes left in the crystals after removing the polymer nanoparticles by dissolution or calcination generate a porous “Swiss Cheese” structure.
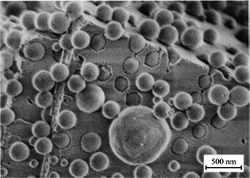 | ||
| Fig. 3 Calcite crystal grown in the presence of carboxyl-functionalized polystyrene latex particles. Latex particles are engulfed in the inorganic crystal. (Reproduced from ref. 18 with kind permission from Springer Science and Business Media.) | ||
The studies with zinc oxide, used as a simple model system with only one hexagonal crystal phase under normal conditions (zincite), demonstrated that negatively charged colloidal particles adsorb specifically on the {001} facets, which retard the growth in the [100] direction. Thus, the elongated hexagonal prismatic crystals obtained in the absence of any additive evolve into shorter and wider shapes. The variation of the surface chemistry of the polymer nanoparticles was shown to have a dramatic effect on the final crystal morphology (Fig. 4).16 Although spectroscopic studies indicated that the physical properties of zinc oxide are influenced by the presence of the polymer,22 careful X-ray diffraction analysis proved that the crystal perfection remains essentially undisturbed in spite of the up to 10 wt% of polymer content,23 which seems to be in agreement with the non-classical crystallization concept.
 | ||
| Fig. 4 Zinc oxide crystallized from aqueous media in the presence of latex particles with different surface chemistry: (a) poly(styrene/maleic acid) and (b) poly(styrene/ethylene glycol methacrylate phosphate). The latex has been removed by calcination at 600 °C. Note the holes in the structure left by the colloid particles.16 | ||
For calcium carbonate, and also using carboxyl-functionalized polystyrene particles, Kim et al.20 have studied how the degree of occlusion of the colloidal particles in the crystal strongly depends on the surface chemistry: particles with higher acid content showed a more effective entrapment, which is consistent with the results previously observed for zinc oxide. Very recently, the same research group, headed by Meldrum, has reported artificial calcite single crystals containing up to 13 wt% of 20 nm micelles of the diblock copolymer PSPMA30-b-PDPA47 (poly(hydroxypropyl methacrylate)30-block-poly(2-(diisopropylamino)ethyl methacrylate)47).24 These micelles, claimed by the authors to act as “pseudo-proteins”, are present in the crystallization medium and become occluded in the crystals, which behave as single crystals and are harder than the counterparts synthesized without the polymer micelles. The carboxyl-functionalized micelles were found to adsorb specifically onto the {104} crystal facets. The approach shows that such materials, prepared in the presence of polymeric colloidal entities, are not only interesting as models for biomineralization, but could also be extended to other systems and become a suitable tool to enhance the mechanical properties of organic–inorganic nanocomposites.25
2.2. Polymer colloids as additives for enantioselective crystallization
The discovery of the importance of stereochemistry in biochemical environments dates back to the days of Pasteur; since then, the study of chirality has been a major topic of research in both academia and industry.26 The definition of chirality is rather straightforward: any two objects that are non-superimposable mirror images of one another are chiral and called enantiomers. The resolution of racemic compounds is important in many fields and the resolution of racemic drugs is especially significant because optical isomers of drugs can exhibit different pharmacological profiles in living systems.A resolution of isomers can be achieved by various processes based on physical, chemical and biological techniques.27 Chiral resolution by crystallization in the presence of chiral additives is a common technique. In this sense, chiral polymers, such as double-hydrophilic block copolymers (DHBCs) with chiral moieties, have been successfully used for chiral resolution via crystallization from conglomerate systems, as first reported by Mastai and Cölfen.28 New approaches for optical resolution and purification of enantiomers using nanosized systems and colloids have been employed.29–31 The use of colloid particles is commonly based on the strategy of linking chiral compounds to the surface and using the resultant material to selectively bind one enantiomer.
In this part, we will review the recent research on colloidal systems as controlling agents for chiral resolution by crystallization.
Although chiral polymers in solution are important for chiral resolution, the use of colloidal particles has attracted much attention in recent years. The application of micron- and submicron-sized chiral colloidal particles for enantioselective crystallization processes provides a very high chiral surface area on which crystallization is favored. Chiral colloidal particles act as resolving auxiliaries in the crystallization of enantiomers. The basic principle of the approach is that enantiospecific colloidal chiral particles will serve as selective chiral nuclei (seeds) during crystallization, lowering the formation energy for crystals of one enantiomer. Consequently, this enantiomer will crystallize in excess on the colloidal chiral particles, thereby enabling the separation of this enantiomer from the crystallization solution. This new chiral resolution technique required the development of new synthetic methods for the preparation of the particles.
Colloidal chiral particles, which can be synthesized by many techniques, are mainly used as a solid support for chiral separation by chromatographic methods. Two of the most prominent methods to prepare colloidal chiral particles are molecular chiral imprinting and the chiral coating of silica or polymer particles with chiral species. Recently, Melamed et al.32 have suggested a new approach for the synthesis of poly(N-vinyl-α-phenylalanine) chiral microspheres. By an analogous method, Menahem et al.33 polymerized L- and D-phenylalanine acryl monomers by a dispersion polymerization procedure and the formed micrometer-sized particles were used as additives in chiral crystallization experiments of amino acid racemates (DL-histidine and DL-glutamic acid) and conglomerate systems (DL-asparagine hydrate, DL-threonine and DL-methionine). In the crystallization of the conglomerate system DL-threonine, the presence of the chiral poly(L-phenylalanine) microspheres showed significant chiral recognition. Scanning electron microscopy demonstrated that crystals were grown on the surface of the chiral particles with significant changes in morphology.
A further example of the preparation of colloidal chiral particles by surface functionalization and their use as controlling agents for chiral crystallization was reported by Krattiger et al.34 In this study, split-and-mix chiral oligopeptide libraries from one to four amino acids were bound to functionalized polystyrene beads. Different oligopeptide sequences were found to be active in the morphogenesis of CaCO3 and DL-alanine. As shown in Fig. 5, the colloidal chiral peptide-functionalized particles serve as selective seeds for the crystallization of DL-alanine, which crystallizes on their surfaces with unique and unusual morphologies. Chiral separation tests for the DL-alanine system indicated that no enantioselective crystallization could be achieved to a significant extent with the applied oligopeptide libraries.
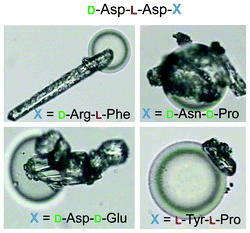 | ||
| Fig. 5 Light microscopy images of peptide-functionalized polystyrene particles (with the corresponding amino acid sequences) and crystal morphogenesis of DL-alanine. (Reprinted from ref. 34, Copyright 2010, with permission from Elsevier.) | ||
Chiral colloidal particles are usually prepared by the chiral functionalization of polystyrene particles. Although such chiral particles exhibit chiral recognition properties, a higher chiral surface would be desirable. Medina et al.35 reported a new synthesis route to prepare the next generation of particles that are entirely, not just the surface, chiral. Chiral N-acryl-L-phenylalanine monomers were polymerized via a one-step swelling dispersion polymerization process. In this method, swollen polystyrene particles served as “reaction vessels” and therefore the polystyrene swelling process is the first step. Non-cross-linked polystyrene particles were added to an oil-in-water microemulsion (chlorobromobenzene or chlorobenzene droplets in water). Polystyrene merged with the organic droplets, thus expanding the particles. In the subsequent step, chiral monomers, the initiator and a cross-linker (divinyl benzene) were dissolved in the swollen polystyrene particle template. Polymerization was initiated by increasing the temperature. After polymerization, the polystyrene particles were dissolved with dry acetone. The outcome of this process was remarkably monodisperse microspheres with a diameter of 5.4 μm (±2%) with controlled morphology and porosity (Fig. 6). Colloidal chiral particles with a perfectly defined structure can be obtained by varying the polymerization conditions, such as the swollen solvents and monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cross-linker ratio. The colloidal particles were used to test the chiral selectivity by enantiomeric adsorption from racemic and enantiomeric pure D- and L-valine solutions. The polymeric particles showed chiral selective adsorption toward the L-valine enantiomer in time-resolved polarimetry experiments. Crystallization experiments of racemic DL-valine in the presence of the chiral particles resulted in enantioselective crystallization on the chiral hollow particles, as shown in Fig. 6(d)–(f). An enantiomeric excess of ca. 25% could be achieved for the crystallization onto these chiral microspheres.
:cross-linker ratio. The colloidal particles were used to test the chiral selectivity by enantiomeric adsorption from racemic and enantiomeric pure D- and L-valine solutions. The polymeric particles showed chiral selective adsorption toward the L-valine enantiomer in time-resolved polarimetry experiments. Crystallization experiments of racemic DL-valine in the presence of the chiral particles resulted in enantioselective crystallization on the chiral hollow particles, as shown in Fig. 6(d)–(f). An enantiomeric excess of ca. 25% could be achieved for the crystallization onto these chiral microspheres.
 | ||
| Fig. 6 (a–c) SEM images of poly(L-phenylalanine) microspheres obtained at different polymerization conditions: (a) hollow structures, (b) porous particles, (c) rough-surface particles. (d–f) SEM images of chiral PV-L-Phe microspheres after crystallization of DL-valine (scale bars = 5 μm).35 | ||
All the examples above describe the preparation and use of micrometer-sized colloidal chiral particles. The synthesis and application of nanosized colloidal chiral particles is, however, more challenging. An example of the preparation of nanosized chiral particles was recently reported by Chen et al.,36 who prepared a novel category of hollow “organic@inorganic” hybrid two-layered nanoparticles in which the inner layer is formed by optically active helical polyacetylene and the outer layer by silica. The one-pot preparation of the hollow chiral nanoparticles consists of three major steps: (i) vinyl polymer cores are formed via free-radical polymerization of the monomer; (ii) middle-shells are formed via aqueous catalytic microemulsion polymerization of the substituted acetylene monomer; and (iii) outer shells of silica are formed by a sol–gel approach. These chiral nanoparticles showed remarkable optical activity and were used as chiral templates to induce the enantioselective crystallization of racemic alanine.
Chiral micellar systems also find application in enantioselective crystallization. The first example of the use of a micellar system for a chiral application was reported by Menahem et al.37 The approach is based on the crystallization of chiral molecules in the aqueous phase of an inverse (water-in-oil) miniemulsion. The chirality in this reverse miniemulsion system is introduced by the use of a chiral amino acid surfactant, based on a N-stearoyl acid with a head group of D- or L-serine. The surfactant molecules are arranged at the water–oil interface, forming a “chiral nanoreactor”. This work demonstrated that the crystallization of chiral systems of calcium tartrate tetrahydrate within these chiral miniemulsion droplets leads to enantioselective crystallization.
3. Preparation of crystalline inorganic colloids by wet-chemical methods: state of the art
Among crystalline colloids, inorganic colloids are particularly exciting due to their manifold functional properties, including optical, electronic, magnetic and catalytic properties.38–40 In the first period of the “colloid science renaissance”, the preparation of monodispersed inorganic colloids, defined as a task between “art and science” by Matijević,41 was based on relatively easy routes, such as controlled precipitation from a homogeneous solution, forced hydrolysis, controlled release of anions or phase transformation (e.g., sol–gel).In the meantime, colloid science has evolved and an impressive variety of synthetic approaches to obtain crystalline inorganic colloids has been described. In this section, we will give an overview of state of the art crystalline inorganic colloids and their preparation routes by liquid-phase methods.39
As stated in the Introduction, we will focus only on wet-chemistry and not consider vacuum-, plasma-, spray-pyrolysis- and ablation-based approaches. We do not intend to provide a comprehensive coverage of the topic, but rather to outline the functional properties that can be enhanced by a controlled and ordered growth of these structures. Furthermore, the overview will be limited to colloids whose nucleation and growth occurs in solution or in suspension.
Wet-chemical routes are chosen for the preparation of inorganic crystalline colloids because the liquid phase is more versatile with respect to the variation of structural, compositional and morphological features of the final materials. Among these methods, aqueous42 and non-aqueous43 sol–gel processes, hydrothermal and solvothermal syntheses44,45 and biomimetic approaches46 will be included in the following discussion. A further distinction between surfactant-assisted and surfactant-free approaches should be done, the latter being characterized by the presence of coordinating solvents that also play the role of stabilizing agents. The pros and cons of surfactant-directed routes with respect to surfactant-free ones have been excellently outlined by Pinna and Niederberger,47 who have also pointed out that organic compounds not only act as an oxygen source during the formation of oxidic nanomaterials, but also strongly affect the size, shape, surface and assembly properties of the crystallites and, in some cases, also the composition and crystal structure.
Inorganic nanocrystals are systems of paramount relevance in modern science and technology and, in particular, in the preparation of functional (nano)materials for advanced applications.48 In this framework, the practical and theoretical mastery of nanocrystal nucleation and growth represents a primary challenge in materials chemistry and in the applications of these systems. One should consider the pivotal role played by the structure, shape and exposed facets of an inorganic nanocrystal in catalysis and optics (e.g., the output of surface-enhanced Raman spectroscopy measurements strongly depends on the structure and facets). Furthermore, the formation of an ordered array of atoms or molecules (i.e., the obtainment of a crystalline structure) is in many cases a condition sine qua non to afford enhanced functional properties. It is, for instance, well known that the photocatalytic properties of titania nanoparticles can be tailored by controlling their crystalline structure and morphology.49,50 Polarz51 has also pointed out the strong effects that shape and morphology have not only on the catalytic properties but also on optical, electronic, mechanical and self-assembly properties. Therefore, the tailoring of particle shape, size, morphology and crystallinity becomes a condition to achieve the desired functionality and properties.
We have referred in Section 2 to the effect of impurities and additives on the growth by adsorption on selective sites. In this context, models predicting the nucleation and growth phenomena can contribute to devise more rational approaches to the preparation of control systems, avoiding time-consuming trial-and-error approaches. Jensen and coworkers52 have developed a relatively simple kinetic model taking into account additional parameters, such as temperature variation or the addition of additives. The model describes the synergetic effects of nanocrystals nucleation and growth and is based on the assumption that monomers assemble to give a larger structure. Actually, the kinetics of the crystal growth of inorganic semiconductor colloids has been the topic of several investigations.53–55 The molecular mechanism of nucleation, the evolution from nuclei to seeds and from seeds to nanocrystals and the influence of thermodynamic and kinetic control have been described by Xia et al.56 in a recent review devoted to the shape-controlled synthesis of metal nanocrystals.
Several synthetic approaches to obtain crystalline nanoparticles, either metallic or binary (oxides, chalcogenides, halides) have been extensively described in books and reviews.39,48,57,58 One of these publications by Dahl et al.59 has focused on “greener approaches” to nanostructured engineered nanomaterials, outlining the possibility of producing ligand-functionalized inorganic nanoparticles in a sustainable, safer and environmentally friendly approach. In this regard, the use of novel dispersing phases, such as ionic liquid and supercritical fluids, has also been reported.60
3.1. Routes to prepare colloidal metal nanoparticles
Metallic nanocrystallites, ranging from large polyatomic clusters (such as Au55) to bigger colloids,58 can be generated either by the dispersion of larger particles (dispersion methods) or by the condensation of smaller units, typically by reducing metal salts in solution in the presence of a stabilizing agent (reduction methods). A typical benchmark among reduction methods is the citrate reduction, pioneered by Turkevich62 over half a century ago, yielding nearly monodisperse, water-soluble gold clusters with diameters ranging from 7 to 100 nm. Gold sols have been the focus of further work.63,64 The well-known citrate method has very high efficiency, since the reduction agent (citrate anion) also acts as a stabilizing ligand for the growing particles. A variation on this pathway is the direct synthesis of ligand-stabilized nanoparticles. A wide range of metal-based nanomaterials can be generated by reducing metal ions in the presence of a capping agent, such as thiols or phosphine groups.58,59Another approach is the so-called germ-growth method, in which the growth starts from smaller colloids and takes place under chemically controlled conditions.61 In fact, good control of the particle shape and size can be pursued by the seeded growth of nanoparticles from smaller seed particles through the epitaxial addition of metal atoms. The seeded growth of nanoparticles utilizes a growth solution that, taking gold nanoparticles as an example, consists of a gold salt, a weak reducing agent and often a surfactant. By this approach, spherical particles, but also anisotropic metal nanoparticles featuring unique optical properties, can be obtained.
Sonochemical methods are a further possibility to generate inorganic colloids. The ultrasounds induce cavitation phenomena, which are associated with several effects of both a physical (microstreaming, mass transport, local heating) and chemical nature (formation of radicals). Several colloids have been sonochemically synthesized in either aqueous or non-aqueous media, including metals (Au, Ag, Pd, Pt, Cu), metal alloys, oxides, chalcogenides and carbides.38 Microwave irradiation can also be used in the synthesis of crystalline nanoparticles. Often used in combination with other synthesis routes (e.g., non-aqueous sol–gel routes43,65), microwave irradiation can lead to highly crystalline materials with reduced time and energy consumption.59
3.2. Routes to prepare colloidal nanoparticles of binary metal compounds (oxides, chalcogenides and halides)
The wet synthesis of metal oxides and other inorganic crystalline colloids is typically based on different approaches, encompassing reactions between the precursors (typically a metal salt and an anion), precipitation, thermal/photoactivated or chemically promoted decomposition of suitable precursors, hydrolysis and condensation and nucleation and growth in a confined space.Binary metal compounds in the form of colloids are very often prepared through hydrothermal or solvothermal routes, in which the precipitation (and crystallization) of the inorganic material takes place in a closed system containing a solution of the precursor at a temperature generally exceeding the boiling point of the solvent and under high pressure. The high pressures involved cause significant changes in the solvent properties, making possible for crystalline structures to form at relatively low temperatures.44,45,66
Many of the wet-chemistry approaches require the presence of organic molecules, which have very diverse functions.65 In addition to the well-known use of surfactants as stabilizers and structure-directing agents to control the size and shape,67 organic molecules can also play unusual roles. For example, Demir et al.68 reported the use of p-toluene sulfonic acid monohydrate as a catalyst for the esterification of zinc acetate to produce ZnO nanocrystals by a solvothermal method.
Surfactant-directed approaches also encompass the aqueous synthesis of metal oxide sols and their subsequent injection into a surfactant solution or the room-temperature decomposition of organometallic precursors in surfactant solutions using the moisture in air or, at higher temperature, using a stabilizing agent, such as oleic acid.69
An alternative “hybrid” route relies on the use of metallomesogens (i.e., metal-containing amphiphiles) as metal oxide precursors instead of a metal species and surfactants.70 The use of ionic liquids as organic solvents constitutes another fast developing research field, which enables the synthesis of various inorganic nanoparticles and nanostructures either in the presence or under the exclusion of water.60,71–73
Among colloidal inorganic systems, increasing attention has been devoted in the last two decades to semiconductors, typically metal oxides or sulfides. Thanks to their tunable optical properties, colloidal semiconductor nanocrystals have gained attention in applications such as electroluminescent devices, bioimaging, microelectronics, catalysis, optics and photonics.74–78
Nanoscale colloidal semiconductor structures with at least one dimension small enough to experience quantum confinement effects have captured the imagination and attention of scientists interested in controlling various chemical and photophysical processes.57,74–78 Aside from having a desirable quantum confinement, colloidal nanocrystals are attractive because they are often synthesized at low temperature, at low cost and with potentially scalable methods using simple wet-chemical reaction batches. The synthesis of these structures, generally referred to as “quantum dots”, is in many cases based on the use of stabilizing surface ligands.
During the growth of nanocrystals, kinetic factors can become more important than thermodynamic ones and, therefore, the binding strength and the time constant at which ligands self-exchange at the surface affect the growth rates of individual facets. Surfaces to which ligands are strongly bound exhibit hindered growth in their respective crystal directions. As a result, the addition of various surface-selective ligands to the reaction mixture alters the growth kinetics along different axes, enabling the control of the nanocrystal shape.79
In 1993, Bawendi and coworkers80 reported the preparation of nanocrystalline cadmium chalcogenides with a narrow size distribution using the “hot injection” method, a landmark work in the colloid-chemistry routes for inorganic nanocrystals based on the fast injection of a suitable reactive precursor into a coordinative solvent, producing a supersaturated solution in which nucleation and growth occur in a controlled way. After this seminal work, numerous new routes were explored and implemented for the synthesis of semiconductor nanoparticles. A well-explored synthetic pathway to produce inorganic semiconductor nanocrystals is based on growth in hot organic surfactants, often also encompassing the role of the solvent.59 Alivisatos et al.74 have outline the pivotal role played by not only the nature of the precursor, able to rapidly decompose at the growth temperature, but also by the organic–inorganic interface and by the ability to use the surface solvation dynamic in a useful fashion.
A very versatile and effective approach to crystalline oxide colloids is the surfactant-free non-aqueous (and/or non-hydrolytic) sol–gel route pioneered by Niederberger and coworkers,43,47,81,82 who have obtained a plethora of nanocrystalline metal oxides, MxOy (M = Ti, Hf, Zr, V, Nb, Mn, Sn, In, Re, Ga, Cu, Al, Ce, Zn, W, La, Ni, etc.), with high compositional homogeneity and purity. The crystallization is conducted at temperatures of 200–300 °C and highly crystalline materials with small crystallite size (5–20 nm) are obtained. In several cases, the ligand-directed assembly of nanocrystals into highly anisotropic nanostructures have been observed. The approach, assessed in the meantime as a valuable alternative to surfactant-assisted and to traditional aqueous sol–gel chemistry routes, relies on quite straightforward synthesis protocols involving only metal oxide precursors and common organic solvents. The obtained uniform nanocrystals exhibit an immense variety of sizes and shapes. Along with this method, the thermal decomposition of metalloorganic precursors and/or oxoclusters or sulfide clusters has also pursued crystalline metal oxide and sulfide nanocrystals with uniform size and narrow size distribution.58,59
Another chemically elegant approach worth mentioning is the polyol-assisted synthesis of nanoscale metal oxides, carbonates, sulfides and halides, exploiting the coordination power and the high viscosity of polyols to limit the growth of the particles.83–85
3.3. Bioinspired approaches
By mimicking the synthetic mechanisms of nature, shape-controlled inorganic nanocrystals can also be obtained using biomolecules as precursors or seeds for growth. Aminoacids, proteins, polysaccharides, lipids and peptides are among the most studied organic biomolecules for controlling and orientating crystal growth: they are the biological version of capping agents in the production of nanocrystals.86,87 Proteins isolated from silica-producing organisms have been used to create TiO2 nanoparticles and other crystalline structures.87,88 As an extension of the concept of a surfactant, biologically produced nanoparticles tend to be stable in solution, perhaps due to protein interactions, further suggesting that purification may not be an issue in some cases. Crystal growth can be directed by the use of special proteins. Since proteins have already been proven to control the crystallization of biological structures (e.g., exoskeletons, bone and seashells), genetically modified proteins can selectively bind to specific crystal faces, so directing the growth.A bioinspired approach to produce inorganic crystalline colloids is the use of multicellular biological organisms, such as bacteria.59 In nature, magnetobacteria are known to be able to synthesize nanosized single crystals of Fe3O4 and FeS2 folded into ordered chain or ring structures and sulfate-reducing bacteria can produce ZnS biofilms under anaerobic conditions. Imitating nature, bacteria and fungi have been applied to produce different inorganic nanoparticles either on the cell surface or intracellularly.89–93
4. Colloidal systems as supports, templates and nanoreactors for inorganic crystallization
It has been shown in Section 2 that colloidal systems, especially colloidal particles, can be used as additives to control the nucleation and growth of crystals. The difference in size between the colloids acting as additives (controlling agents) and the grown crystals is typically at least one order of magnitude, the former being <1 μm and the latter in the range of several micrometers. Much more commonly, however, colloidal systems are applied as supports or templates for crystallization, and these methods will be discussed in the following section.Roughly speaking, two main approaches can be distinguished: (i) hard templating, in which colloidal particles (typically silica or polymer particles) act as a support for the formation of crystals on the surface, and (ii) soft templating, in which colloidal structures (e.g., micelles) confine crystallization, mostly in emulsion and phase separation procedures. This distinction between “hard” and “soft” templates, schematically represented in Fig. 7, originally comes from the area of hollow particle preparation,94–96 but it can be generalized to other template crystallization processes happening in the presence of colloidal systems. Aiming to focus on crystallization from solution, we will not describe the long list of works in which crystallization occurs in the solid state after calcination of amorphous materials produced by sol–gel methods.
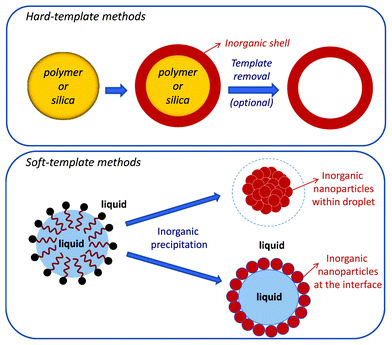 | ||
| Fig. 7 Schematic representation of hard and soft templating. | ||
4.1. Crystallization on unfunctionalized colloidal particles: direct deposition methods
Already in the early 1990s, before the preparation of core–shell hybrid particles had become a hot topic, Matijević and coworkers did some pioneering work and reported the precipitation of inorganic materials, such as yttrium basic carbonate (Y(OH)CO3)97 and some zirconium compounds (Zr2O2(OH)2CO3, Zr2 (OH)6SO4),98 on the surface of simple polystyrene particles, cationically or anionically stabilized, but with no further treatment or specific functionalization. There are also a few reported examples of the direct crystallization of metals and metal oxides either on silica or on polymer colloids. Zhang et al.99 showed that silver or metal nanoparticles can be precipitated on the surface of polystyrene and silica beads, making use of the electrostatic attraction between positively charged metal complexes and the preexisting negative charges on the surface of the colloids, arising from the surfactant used in the synthesis of the latex and from the –OH groups in the case of the silica particles. Huang et al.100 reported the precipitation of Fe3O4 on the surface of sulfate-stabilized polystyrene latex particles in the presence of ethylene glycol. Lynden and coworkers also demonstrated a few years ago that tin(IV) oxide can be directly deposited on the surface of silica nanospheres101 or even on silica-coated α-Fe2O3 spindles, resulting in fancy “double-walled SnO2 nano-cocoons” (Fig. 8).102 The as-prepared nanocrystals are tetragonal rutile SnO2, although a pronounced peak broadening of the X-ray diffraction peaks is reported, which is coherent with the small crystallite sizes obtained.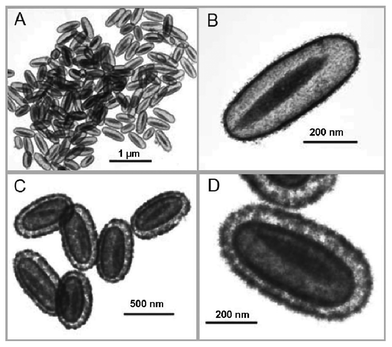 | ||
| Fig. 8 TEM images of single-walled SnO2 nano-cocoons with movable α-Fe2O3 spindles and porous double-shelled SnO2 nano-cocoons. (Reproduced from ref. 102 with permission from John Wiley & Sons.) | ||
Another efficient strategy to obtain crystalline direct deposition from solution on the surface of colloidal particles is the application of ultrasound. Gedanken and coworkers have made very significant contributions and have successfully crystallized in situ different inorganic materials on the surface of colloidal particles, including ZnS,103 metallic cobalt,104 gold on silica105 and metallic nickel on alumina.106
4.2. Crystallization on particles coated with adsorbed polyelectrolytes: conventional hard-template methods
As indicated above, the use of hard templates has been a conventional way to obtain inorganic hollow particles, very often by applying the “layer-by-layer” (LbL) deposition technique. Characterized by its simplicity, effectivity and versatility, many research groups, such as those of Möhwald and Caruso, have extensively used this approach and the advances in the topic have been summarized in numerous review papers.94,107–109 As suggested by the name, the LbL technique involves the sequential deposition of oppositely charged polyelectrolytes on the supporting particle. A common option is to prepare the crystalline nanoparticles ex situ and deposit them afterwards on the surface of template beads layered with polyelectrolytes.108–111 An alternative strategy is the in situ crystallization of the nanoparticles on the surface of the templates, but the LbL technique presents important limitations in that sense because the addition of precipitating agents required for the in situ reactions can destabilize the deposited layers of polyelectrolytes. This technique has, however, been successful in the preparation of noble metal nanoparticles on the surface of colloids by the reduction of deposited metal precursors. Antipov et al.112 reported the precipitation of silver nanoparticles on the surface of poly(sodium styrenesulfonate)/poly(allylamine hydrochloride) hollow capsules. Similarly, Bruening and coworkers113 prepared Pd nanoparticles on the surface of alumina particles covered with adsorbed polyelectrolytes and Tian et al.114 deposited gold nanoparticles on polystyrene nanoparticles modified with poly(ethyleneimine).Sol–gel precursors can also be adsorbed on colloids covered with polyelectrolytic layers, but the obtained materials are almost always amorphous. For example, Caruso's research group reported the preparation of TiO2115 and LiNbO3116 by depositing the inorganic precursor on the surface of polystyrene particles in combination with the sequential adsorption of poly(diallyl dimethylammonium chloride) and poly(styrenesulfonate), but the crystallization occurred only after the calcination of the materials, which falls beyond the focus of this article.
Also using the LbL deposition technique, Valtchev117 prepared zeolite A crystals on the surface of polystyrene microspheres treated with a cationic polymer, which facilitated the adsorption of negatively charged zeolite A crystal seeds. These seeds on the surface served as nucleation sites for the subsequent growth.
4.3. Crystallization on covalently functionalized polymer particles
In the examples of particles as supports (or hard templates) presented so far, the colloidal core plays mostly a sacrificial role and the functional groups on the surface are either “intrinsic” (hydroxyl groups on silica) or merely adsorbed (like in the LbL case). A different approach is the use of polymer particles with covalently attached functionalities, which are able to bind metal ions and serve as nucleation centers for crystallization (Fig. 9). As it is not always desirable to remove the core but to exploit the properties of the hybrid materials, it may be more appropriate to speak of a “support” rather than a “hard template”. Depending on the system and on the preparation conditions, the crystals deposited on the particle may not completely cover the surface but form a “raspberry-like” structure, which is not necessarily a drawback but can be an interesting feature. Agrawal et al.118 have reviewed in a recent paper the developments on the fabrication of polymer–inorganic hybrid particles.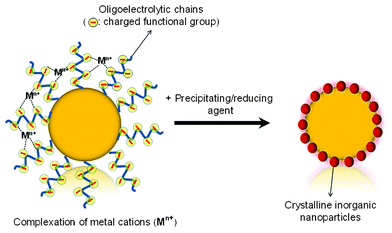 | ||
| Fig. 9 General strategy for the precipitation/crystallization of metal-containing inorganic materials on the surface of functionalized latex particles. | ||
As in the previous strategies, a big portion of the accomplished work has been for noble metal nanoparticles, which are readily precipitated by the reduction of metal salts. Ballauff's research group has been one of the most active in this topic and has reported the formation of Ag, Au, Pd and Pt nanoparticles on the surface of “hairy” latexes, functionalized with long polyelectrolytic chains.119–123 In a recent work, Lu and coworkers have reported the growth of Pt/Au nanorods on poly(N-isopropylacrylamide) microgels attached to a polystyrene core.129 Other groups have reported analogous results.125–127 It is worth noting that both amorphous and crystalline noble metal nanoparticles can be obtained, depending on the size of the nanoparticles. According to Schrinner et al.,128 gold particles larger than 2 nm are crystalline, while those in the range of 1.3 nm are amorphous—at least “X-ray amorphous”.
Because of the importance in biomineralization and biomedical applications, hydroxyapatite, Ca5(PO4)3(OH), is another widely studied crystalline material. More than ten years ago, Tamai and Yasuda129 already presented the formation of hydroxyapatite on poly(styrene-co-acrylic acid) latex prepared by emulsion polymerization. Similar latex particles, although prepared by miniemulsion, were used as a support for the crystallization by Ethirajan et al.130,131 Hydroxyapatite has also been crystallized on polystyrene particles with other functionalities, resulting from the copolymerization of styrene with monomers such as acetoacetoxyethylmethacrylate (AAEMA),132 vinylphosphonic acid (VPA) and vinylbenzylphosphonic acid (VBPA).133
The use of ionic surfactants and initiators like potassium persulfate in emulsion polymerization techniques leads to the presence of charge groups on the surface, which can interfere with crystallization processes. Even non-ionic surfactants, such as Lutensol AT50, a commercially available hexadecyl-modified poly(ethylene oxide) with ca. 50 ethylene oxide units, can also have a negative effect on crystallization if the concentration is too high.131 Therefore, for better control of the crystallization on the surface of polymer particles, a convenient alternative to achieve functionalization is the use of surface-active monomers (so-called “surfmers”) containing the desired functionality. Surfmers act simultaneously as monomers and surfactants. This approach avoids the use of an additional surfactant and the surfmer copolymerize with the monomer of the core, becoming covalently bound to the surface. Recent works have reported the crystallization of hydroxyapatite on the surface of particles prepared with a phosphonate surfmer134 and the use of phosphate and phosphonate surfmers to prepare functionalized particles for the crystallization of different metal oxides.135
The successful crystallization of metal oxides, hydroxides and chalcogenides on the surface of polymer particles is more complex than for the case of metals and calcium phosphates. Poly(styrene-co-acetoxyethyl methacrylate) latex particles prepared with potassium persulfate as an initiator have been used for the in situ crystallization of ZnO and In(OH)3 from 2-propanol136,137 and ZnS from aqueous media.138 Analogous experiments for TiO2139 and Ta2O5140 resulted in amorphous materials unless calcinated. Very remarkably, Lu et al.141 showed that TiO2 could be obtained at room temperature as crystalline anatase on the surface of polystyrene particles functionalized with poly(styrene sodium sulfonate) with no need for any further thermal treatment. These results are significant because TiO2 prepared by sol–gel is mostly amorphous.
A recent work by Fischer et al.135 has shown that phosphate and phosphonate functionalities, which originate from copolymerization with specifically synthesized surfmers, are very suited for the controlled surface precipitation of CeO2, α-Fe2O3, Fe3O4 and ZnO from both aqueous and alcoholic media. Interestingly, ZnO could be obtained in a crystalline form at room temperature from alcoholic media but not from aqueous solutions. In all the systems studied, the initial polymer dispersions coagulated and became unstable after adding the metal precursor but were observed to be stable again after the addition of the precipitating agent and the formation of the crystals (Fig. 10).
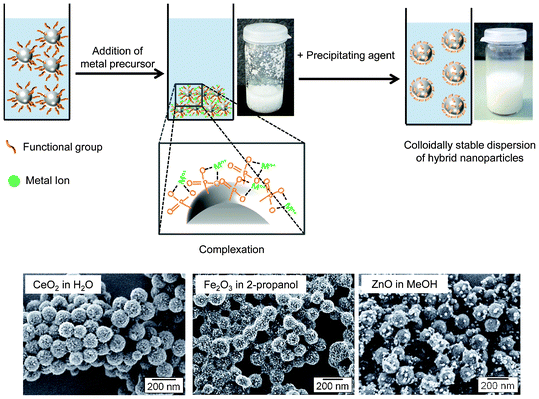 | ||
| Fig. 10 Formation of metal oxide nanoparticles on the surface of phosphonate-functionalized polymer particles: schematic representation of the process and SEM images of different metal oxide–polymer hybrid particles. (Adapted from ref. 135. Reproduced with permission from John Wiley & Sons.) | ||
As an overview of the work conducted in the last few years in the area, Table 1 summarizes some representative publications on the crystallization of inorganic nanoparticles on the surface of covalently functionalized polymer particles, indicating the crystallization solvent and the specific surface chemistry.
| Nanocrystalline material | Crystallization solvent | Colloidal polymer supporta | Reference(s) |
|---|---|---|---|
| a S: styrene, NIPAM: N-isopropylacrylamide, AEMH: 2-aminoethylmethacrylate, AA: acrylic acid, AAEMA: acetoacetoxyethylmethacrylate, VPA: vinylphosphonic acid, VBPA: vinylbenzylphosphonic acid, SS: styrene sodium sulfonate. | |||
| Ag | Water | Poly(S/NIPAM) | 119,126 |
| Au | Water | Poly(S/AEMH) | 123 |
| Au/Pt | Water | Poly(S/NIPAM) | 124 |
| Ca5(PO4)3(OH) | Water | Poly(S/AA) | 129–131 |
| Ca5(PO4)3(OH) | Water | Poly(S/AAEMA) | 132 |
| Ca5(PO4)3(OH) | Water | Poly(S/VPA) or poly(S/VBPA) | 133 |
| Ca5(PO4)3(OH) | Water | Poly(S/R–PO3H2) | 134 |
| CeO2 | Water | Poly(S/R–PO3H2) or poly(S/R–PO4H2) | 135 |
| Fe2O3 | Water, 2-propanol | Poly(S/R–PO3H2) or poly(S/R–PO4H2) | 135 |
| Fe3O4 | Water | Poly(S/RPO3H2) or poly(S/RPO4H2) | 135 |
| In(OH)3 | 2-propanol | Poly(S/AAEMA) | 142 |
| Pd | Water | Poly(S/R–NH2) | 127 |
| Pd | Water | Poly(S/R–N(CH3)3Cl) | 120 |
| Pt | Ethanol–water | Poly(S/NIPAM) | 125 |
| Pt | Water | Poly(S/R–(CH3)3NCl) | 121 |
| Pt | Water | Poly(S/AEMH) | 122 |
| TiO2 | Ethanol | Poly(S/SS) | 141 |
| ZnO | 2-propanol | Poly(S/AAEMA) | 136 |
| ZnO | Methanol, 2-propanol or ethanol | Poly(S/RPO3H2) or poly(S/RPO4H2) | 135 |
| ZnO/TiO2 | 2-Propanol–ethanol | Poly(S/AAEMA) | 137 |
| ZnS | Water | Poly(S/AAEMA) | 138 |
4.4. Microgel particles as scaffolds for crystallization
Some of the examples mentioned in the previous subsection, like those using polyacrylamides to functionalize “hard” polystyrene spheres, involve the interaction of microgels with metal ions. It is also possible, however, to apply particles made only of the “soft” microgel (i.e., without any “hard” core) for precipitation and crystallization processes. It can be considered that such materials are, somehow, halfway between hard and soft templates. The applications of hydrogel nanoparticles for the synthesis of new materials has been reviewed in different articles.143–145Antonietti et al.146 were the first to demonstrate the use of microgel particles as nanoreactors for inorganic nanoparticle synthesis and they reported the preparation of noble metal colloids templated with poly(styrene sulfonate) gels. Some years later, Kumacheva and coworkers147 used poly(N-isopropyl acrylamide/acrylic acid/2-hydroxyethylacrylate) microgels to prepare metal nanoparticles as well, but they also extended the approach to CdS. In the last few years, microgel (and nanogel) particles of different chemistries have been used as scaffolds for the preparation of a variety of crystalline materials, including CaCO3,148 ZnS,138 calcium phosphates and hydroxyapatite149–151 and Bi2O3.152
4.5. Soft-template methods and crystallization within “colloidal nanoreactors”
While hard templates (or supports) are solid colloidal particles, soft templates typically involve structures and spaces confined by amphiphilic molecules, such as surfactants or amphiphilic polymers. This includes emulsion droplets (micro- and miniemulsion droplets), micelles, micellar aggregates and vesicles. The structures generated by soft templates confine the spaces in which crystallization can take place, that is, these spaces act as “nanoreactors” and allow the control of both the nucleation and growth of crystalline materials due to the confinement of the crystallization process. This “nanoreactor” concept discloses exciting perspectives for the morphsynthetic control of the crystalline habit of the formed structures. In fact, the physical properties of liquids in nanodroplets can be substantially different from those of the bulk phase and the dynamics of the crystallization and melting process are consequently affected. In particular, in both direct and inverse emulsion systems, the temperature of crystallization is significantly decreased with respect to the bulk phase because heterogeneous nucleation is practically suppressed.Different from microemulsions, which are thermodynamically stable systems, miniemulsions are kinetically stabilized and the concentration of surfactant stays under the critical micellar concentration, so that it is not appropriate to speak about micelles, but rather about “stabilized droplets”. Miniemulsions are obtained by high energy homogenization, typically induced by ultrasound, yielding very fine dispersions. Especially in the food industry,156 but also occasionally in colloid chemistry,157,158 naming the miniemulsions with the younger term ‘nanoemulsions’ is starting to became popular. A look at the publications indexed by the Web of Knowledge shows that ‘miniemulsion’, coined by the emulsion polymerization community, dates back to at least 1984, while the term ‘nanoemulsion’ appears for the first time in an indexed publication as late as 1996. From our side, we encourage the term ‘miniemulsion’ not only because of historical reasons, but also because ‘nanoemulsion’ (or ‘nano-emulsion’ with a hyphen, as spelled by some authors158) may be misleading regarding the size of the droplets when compared with ‘microemulsion’.
For the synthesis of inorganic particles, most commonly, two emulsions containing the precursors (or a reducing/precipitating agent) are combined, so that the precipitation reaction occurs after the collision and coalescence of the droplets containing the reactants (Fig. 11). If only one emulsion is used and the precipitating agent is present or added into the continuous phase, the precipitation may occur at the droplet liquid–liquid interface and hollow particles are formed. Most of the materials produced in this way at the interface by sol–gel are amorphous and crystallize only upon thermal treatment. However, crystalline materials have also been produced in macro-, mini- and microemulsions. Micron- and submicron-sized hollow spheres of metals (Ni, Pt),159,160 oxides (MnO2)161 and chalcogenides (ZnS, CuS2 and CdS)162–164 have been reported, to mention some examples.
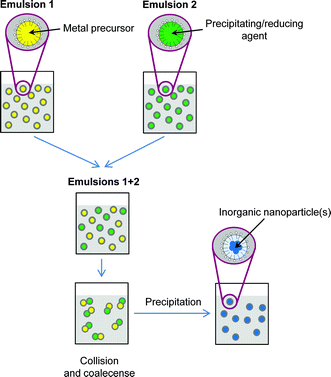 | ||
| Fig. 11 Scheme representing the formation of nanoparticles by the combination of two micro- or miniemulsions. | ||
The use of microemulsions in nanoparticle synthesis has been reviewed in different papers.165–167 Especially interesting in obtaining hollow particles by microemulsions are the achievements of Feldmann and coworkers, who have reported nanocrystalline hollow nanospheres of different oxides and hydroxides, including γ-AlO(OH),168 La(OH)3,169 SnO2170 and TiO2.171 Although the TiO2 obtained appeared to be amorphous according to the X-ray diffraction data, high-resolution transmission electron microscopy showed that the shell of the capsules was, at least partially, crystalline.171 Another research group has also prepared zirconium phosphates172 and hydrotalcites173,174 in a microemulsion. Although inorganic nanoparticles are mostly prepared in inverse (oil-in-water) systems, direct microemulsions have also been used. Ge et al.175 reported the formation of a large variety of inorganic nanoparticles at the oil–water interface in microemulsion droplets. Some of the nanoparticles (Ag2S, PbS, Ag2Se, PbSe, Ag, Cu) were shown to be crystalline. Sánchez-Domínguez et al.176–178 synthesized metallic (Pt, Pd, Rh) and metal oxide (CeO2, ZrO2, Ce0.5Zr0.5O2, TiO2) nanoparticles under mild conditions starting from organometallic precursors dissolved in the oil dispersed phase. In a very recent study, Okoli et al.179 have compared the properties of magnetic iron oxide nanoparticles obtained in oil-in-water and water-in-oil microemulsions.
The application of miniemulsions to the synthesis of inorganic nanoparticles, although not exclusively focused on crystallization processes, has been very recently reviewed elsewhere180 and we will discuss it here only briefly. In the first reported inorganic synthesis in a miniemulsion, Willert et al.181 demonstrated in the early 2000s the versatility of the miniemulsion technique and reported the crystallization of calcium carbonate and iron(III) oxide. The recrystallization of molten salts (zirconium oxychloride) and metals (gallium and alloys) was also achieved by decreasing the temperature below the melting point so that the molten substance solidified. Taden et al.182 also reported the preparation of complex lanthanide phosphors with the composition (Y0.04Eu0.06)2O3, La0.5C0.3Tb0.2PO4 and Ba0.90Eu0.1MgAl10O17. Using inverse (water-in-oil) miniemulsions, TiO2,183 Ce2O184 and Ti1−xZrxO2 (0.005 ≤ x ≤ 0.5)185 have been obtained. Recently, Dolcet et al.186 observed the crystallization of nanoscopic ZnO already at room temperature by exploiting the precipitation of the oxide in the confined space of the droplets in an inverse miniemulsion. In this case, in basic or neutral media, the formation of the amphoteric hydroxide species, ZnOx(OH)y(OH2)z, would have been expected. Actually, prolonged storing and aging in its mother liquor promotes the formation of the crystalline oxide. The results evidenced that the confined space of a droplet, filled with the mother liquor, favoured the aging.

5. Summary and outlook
The aim of this highlight paper has been to underline that the importance of colloidal systems in crystallization processes goes much beyond conventional nanocrystalline colloids. It has been shown that colloidal particles and micelles can be used as additives to control the nucleation and growth of inorganic crystallization. The materials formed, which occlude the colloidal entities, are not only interesting for their application as models for the understanding of biomineralization processes, but recent studies have shown that the physical properties of the materials are also influenced, opening the door to a new generation of hybrid materials. One of the crucial open questions is the mechanistic understanding of the occlusion process, through which colloidal entities become, or not, embedded on the growing crystal.Colloids can also be used in enantioselective crystallization to promote the growth of a specific chiral compound, allowing for the separation of racemic mixtures. A current challenge in this field is the extension of the results reported so far for microspheres to the nanoscale.
The preparation of inorganic colloids has advanced significantly from the early 1990s till now, in particular on the control of the size and the morphology of the obtained nanocrystals and on tuning the final properties. The recent developments in soft and hard templating for preparing inorganic colloids have been reviewed. Although hard templating is efficient and relatively straightforward, it has some limitations: in the case of hollow structures, it is more difficult to load the capsule with the desired material than in the case of soft templates, especially in the case of liquids. In the last few years, significant progress has been achieved in the crystallization of inorganic matter on the surface of covalently functionalized nanoparticles. In the area of soft templating, a remaining challenge is the difficulty of controlling the particle size of the droplets due to the coalescence of the droplets or nanocontainers carrying the inorganic precursor.
Although, in comparison with the so-called physical methods, crystals produced in the liquid phase may be less pure or have a higher degree of crystal defects, which may be important for certain optical or electronic applications, wet-chemical approaches are not only much simpler, but also much more versatile. Furthermore, as the colloidal crystalline particles are already suspended in a liquid, a chemical postfunctionalization is often possible without additional redispersion steps, being less tedious and time consuming. In this regard, postfunctionalization steps may be required to generate multifunctional colloids, which is one of the trends of current research in the field. Functionalization (a priori or a posteriori) may also be a key issue, combined with the control of the shape and size, towards the self-assembly of colloidal entities.
References
- IUPAC, Compendium of Chemical Terminology: Gold Book, 2012 Search PubMed.
- D. J. Barber and I. C. Freestone, Archaeometry, 1990, 32, 33–45 CrossRef.
- K. An and T. Hyeon, Nano Today, 2009, 4, 359–373 CrossRef CAS.
- N. Gaponik, S. G. Hickey, D. Dorfs, A. L. Rogach and A. Eychmüller, Small, 2010, 6, 1364–1378 CrossRef CAS.
- H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362–1395 CrossRef CAS.
- K. Sangwal, Additives and Crystallization Processes: From Fundamentals to Application, Wiley, New York, 2007 Search PubMed.
- R. Q. Song and H. Cölfen, CrystEngComm, 2011, 13, 1249–1276 RSC.
- H. Cölfen, in Biomineralization II: Mineralization Using Synthetic Polymers and Templates, ed. K. Naka, Topics in Current Chemistry, vol. 271, Springer, Heidelberg, 2007, pp. 1–77 Search PubMed.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS.
- A. W. Xu, Y. R. Ma and H. Cölfen, J. Mater. Chem., 2007, 17, 415–449 RSC.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef.
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- H. Cölfen and M. Antonietti, Mesocrystals and Nonclassical Crystallization, Wiley, West Sussex, 2008 Search PubMed.
- R. J. Davey and J. W. Mullin, J. Cryst. Growth, 1974, 23, 89–94 CrossRef CAS.
- G. Wegner, P. Baum, M. Müller, J. Norwig and K. Landfester, Macromol. Symp., 2001, 175, 349–355 CrossRef CAS.
- R. Muñoz-Espí, Y. Qi, I. Lieberwirth, C. M. Gómez and G. Wegner, Chem.–Eur. J., 2006, 12, 118–129 CrossRef.
- K. Gorna, R. Muñoz-Espí, F. Gröhn and G. Wegner, Macromol. Biosci., 2007, 7, 163–173 CrossRef CAS.
- G. Wegner, M. M. Demir, M. Faatz, K. Gorna, R. Muñoz-Espí, B. Guillemet and F. Gröhn, Macromol. Res., 2007, 15, 95–99 CrossRef CAS.
- C. H. Lu, L. M. Qi, H. L. Cong, X. Y. Wang, J. H. Yang, L. L. Yang, D. Y. Zhang, J. M. Ma and W. X. Cao, Chem. Mater., 2005, 17, 5218–5224 CrossRef CAS.
- Y. Y. Kim, L. Ribeiro, F. Maillot, O. Ward, S. J. Eichhorn and F. C. Meldrum, Adv. Mater., 2010, 22, 2082–2086 CrossRef CAS.
- V. Fischer, K. Landfester and R. Muñoz-Espí, Cryst. Growth Des., 2011, 11, 1880–1890 CAS.
- R. Muñoz-Espí, G. Jeschke, I. Lieberwirth, C. M. Gómez and G. Wegner, J. Phys. Chem. B, 2007, 111, 697–707 CrossRef.
- R. Muñoz-Espí, A. Chandra and G. Wegner, Cryst. Growth Des., 2007, 7, 1584–1589 Search PubMed.
- Y. Y. Kim, K. Ganesan, P. C. Yang, A. N. Kulak, S. Borukhin, S. Pechook, L. Ribeiro, R. Kröger, S. J. Eichhorn, S. P. Armes, B. Pokroy and F. C. Meldrum, Nat. Mater., 2011, 10, 890–896 CrossRef CAS.
- L. A. Estroff and I. Cohen, Nat. Mater., 2011, 10, 810–811 CrossRef CAS.
- T. J. Maher and D. A. Johnson, Drug Dev. Res., 1991, 24, 149–156 CrossRef CAS.
- N. M. Maier, P. Franco and W. Lindner, J. Chromatogr., A, 2001, 906, 3–33 CrossRef CAS.
- Y. Mastai, M. Sedlak, H. Cölfen and M. Antonietti, Chem.–Eur. J., 2002, 8, 2430–2437 CrossRef.
- D. B. Amabilino, Chem. Soc. Rev., 2009, 38, 669–670 RSC.
- D. S. Bag, T. C. Shami and K. U. B. Rao, Def. Sci. J., 2008, 58, 626–635 Search PubMed.
- Y. Mastai, Chem. Soc. Rev., 2009, 38, 772–780 RSC.
- O. Melamed and S. Margel, J. Colloid Interface Sci., 2001, 241, 357–365 CrossRef CAS.
- T. Menahem and Y. Mastai, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3009–3017 CrossRef CAS.
- P. Krattiger, N. Nassif, A. Völkel, Y. Mastai, H. Wennemers and H. Cölfen, Colloids Surf., A, 2010, 354, 218–225 CrossRef CAS.
- D. D. Medina, J. Goldshtein, S. Margel and Y. Mastai, Adv. Funct. Mater., 2007, 17, 944–950 CrossRef CAS.
- B. Chen, J. Deng and W. Yang, Adv. Funct. Mater., 2011, 21, 2345–2350 CrossRef CAS.
- T. Menahem and Y. Mastai, New J. Chem., 2008, 32, 925–928 RSC.
- Colloids and Colloid Assemblies: Synthesis, Modification, Organization and Utilization of Colloid Particles, ed. F. Caruso, Wiley-VCH, Weinheim, 2004 Search PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- S. E. Lohse and C. J. Murphy, J. Am. Chem. Soc., 2012, 134, 15607–15620 CrossRef CAS.
- E. Matijević, Langmuir, 1986, 2, 12–20 CrossRef.
- C. J. Brinker and G. W. Scherer, The Physics and Chemistry of Sol–Gel Processing, Academic Press, San Diego, CA, 1990 Search PubMed.
- I. Bilecka and M. Niederberger, Electrochim. Acta, 2010, 55, 7717–7725 CrossRef CAS.
- K. Byrappa and M. Yoshimura, Handbook of Hydrothermal Technology, Noyes Publications/William Andrew Publishing, Norwich, NY, 2001 Search PubMed.
- U. Schubert and N. Hüsing, Synthesis of Inorganic Materials, Wiley-VCH, Weinheim, 2012 Search PubMed.
- S. Mann and G. A. Ozin, Nature, 1996, 382, 313–318 CrossRef CAS.
- N. Pinna and M. Niederberger, Angew. Chem., Int. Ed., 2008, 47, 5292–5304 CrossRef CAS.
- M. P. Pileni, Surf. Sci., 2009, 603, 1498–1505 CrossRef CAS.
- M. D'Arienzo, J. Carbajo, A. Bahamonde, M. Crippa, S. Polizzi, R. Scotti, L. Wahba and F. Morazzoni, J. Am. Chem. Soc., 2011, 133, 17652–17661 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- S. Polarz, Adv. Funct. Mater., 2011, 21, 3214–3230 CrossRef CAS.
- J. Y. Rempel, M. G. Bawendi and K. F. Jensen, J. Am. Chem. Soc., 2009, 131, 4479–4489 CrossRef CAS.
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16646–16654 CrossRef CAS.
- S. G. Hickey, C. Waurisch, B. Rellinghaus and A. Eychmüller, J. Am. Chem. Soc., 2008, 130, 14978–14980 CrossRef CAS.
- X. Peng, J. Wickham and A. P. Alivisatos, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- Nanoparticles: From Theory to Application, ed. G. Schmid, 2nd edition, Wiley-VCH, 2010 Search PubMed.
- S. G. Kwon and T. Hyeon, Acc. Chem. Res., 2008, 41, 1696–1709 CrossRef CAS.
- J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228–2269 CrossRef CAS.
- M. Antonietti, B. Smarsly and Y. Zhou, in Ionic liquids in synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2008, pp. 609–617 Search PubMed.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- G. Schmid and B. Corain, Eur. J. Inorg. Chem., 2003, 3081–3098 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- G. Garnweitner and M. Niederberger, J. Mater. Chem., 2008, 18, 1171–1182 RSC.
- W. L. Suchanek and R. E. Riman, in Ceramics Science and Technology, Wiley-VCH, Weinheim, 2011, pp. 63–95 Search PubMed.
- Ö. Tunusoglu, R. Muñoz-Espí, Ü. Akbey and M. M. Demir, Colloids Surf., A, 2012, 395, 10–17 CrossRef.
- M. M. Demir, R. Muñoz-Espí, I. Lieberwirth and G. Wegner, J. Mater. Chem., 2006, 16, 2940–2947 RSC.
- M. A. Sliem, D. A. Schmidt, A. Bétard, S. B. Kalidindi, S. Gross, M. Havenith, A. Devi and R. A. Fischer, Chem. Mater., 2012, 24, 4274–4282 CrossRef CAS.
- S. Polarz, R. Regenspurger and J. Hartmann, Angew. Chem., Int. Ed., 2007, 46, 2426–2430 CrossRef CAS.
- A. V. Mudring, T. Alammar, T. Baecker and K. Richter, in Ionic Liquids: From Knowledge to Application, ACS Symp. Ser., Vol. 1030, American Chemical Society, 2009, pp. 177–188 Search PubMed.
- H. Zhang and H. Cui, Langmuir, 2009, 25, 2604–2612 CrossRef CAS.
- D. Freudenmann, S. Wolf, M. Wolff and C. Feldmann, Angew. Chem., Int. Ed., 2011, 50, 11050–11060 CrossRef CAS.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS.
- E. H. Sargent, Nat. Photonics, 2012, 6, 133–135 CrossRef CAS.
- A. L. Rogach, D. V. Talapin and H. Weller, in Colloids and Colloid Assemblies: Synthesis, Modification, Organization and Utilization of Colloid Particles, ed. F. Caruso, Wiley-VCH, Weinheim, 2004, pp. 52–95 Search PubMed.
- W. Li and I. Shah, in Encyclopedia of Nanoscience and Nanotechnology, American Scientific Publishers, 2004, pp. 669–695 Search PubMed.
- A. Shavel, N. Gaponik and A. Eychmüller, Eur. J. Inorg. Chem., 2005, 3613–3623 CrossRef CAS.
- B. K. Hughes, J. M. Luther and M. C. Beard, ACS Nano, 2012, 6, 4573–4579 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS.
- M. Niederberger and N. Pinna, Metal Oxide Nanoparticles in Organic Solvents, Springer, Dordrecht, 2009 Search PubMed.
- C. Feldmann, Adv. Funct. Mater., 2003, 13, 101–107 CrossRef CAS.
- E. Hammarberg, A. Prodi-Schwab and C. Feldmann, J. Colloid Interface Sci., 2009, 334, 29–36 CrossRef CAS.
- P. Schmitt, N. Brem, S. Schunk and C. Feldmann, Adv. Funct. Mater., 2011, 21, 3037–3046 CrossRef CAS.
- P. Curnow, P. H. Bessette, D. Kisailus, M. M. Murr, P. S. Daugherty and D. E. Morse, J. Am. Chem. Soc., 2005, 127, 15749–15755 CrossRef CAS.
- B. L. Smith, G. T. Paloczi, P. K. Hansma and R. P. Levine, J. Cryst. Growth, 2000, 211, 116–121 CrossRef CAS.
- K. Shimizu, J. Cha, G. D. Stucky and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6234–6238 CrossRef CAS.
- R. Y. Sweeney, C. Mao, X. Gao, J. L. Burt, A. M. Belcher, G. Georgiou and B. L. Iverson, Chem. Biol., 2004, 11, 1553–1559 CrossRef CAS.
- A. Ahmad, P. Mukherjee, S. Senapati, D. Mandal, M. I. Khan, R. Kumar and M. Sastry, Colloids Surf., B, 2003, 28, 313–318 CrossRef CAS.
- A. Ahmad, S. Senapati, M. I. Khan, R. Kumar and M. Sastry, Langmuir, 2003, 19, 3550–3553 CrossRef CAS.
- M. Kowshik, N. Deshmukh, W. Vogel, J. Urban, S. K. Kulkarni and K. M. Paknikar, Biotechnol. Bioeng., 2002, 78, 583–588 CrossRef CAS.
- D. J. H. Gaskin, K. Starck and E. N. Vulfson, Biotechnol. Lett., 2000, 22, 1211–1216 CrossRef CAS.
- F. Caruso, Top. Curr. Chem., 2003, 227, 145–168 CrossRef CAS.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2007, 20, 3987–4019 CrossRef.
- J. Liu, F. Liu, K. Gao, J. S. Wu and D. F. Xue, J. Mater. Chem., 2009, 19, 6073–6084 RSC.
- N. Kawahashi and E. Matijević, J. Colloid Interface Sci., 1990, 138, 534–542 CrossRef CAS.
- N. Kawahashi, C. Persson and E. Matijević, J. Mater. Chem., 1991, 1, 577–582 RSC.
- J. H. Zhang, J. B. Liu, S. Z. Wang, P. Zhan, Z. L. Wang and N. B. Ming, Adv. Funct. Mater., 2004, 14, 1089–1096 CrossRef CAS.
- Z. B. Huang, F. Q. Tang and L. Zhang, Thin Solid Films, 2005, 471, 105–112 CrossRef CAS.
- X. W. Lou, C. Yuan and L. A. Archer, Small, 2007, 3, 261–265 CrossRef CAS.
- X. W. Lou, C. Yuan and L. A. Archer, Adv. Mater., 2007, 19, 3328–3332 CrossRef CAS.
- N. A. Dhas, A. Zaban and A. Gedanken, Chem. Mater., 1999, 11, 806–813 CrossRef CAS.
- S. Ramesh, Y. Cohen, R. Prozorov, K. V. P. M. Shafi, D. Aurbach and A. Gedanken, J. Phys. Chem. B, 1998, 102, 10234–10242 CrossRef CAS.
- V. G. Pol, A. Gedanken and J. Calderon-Moreno, Chem. Mater., 2003, 15, 1111–1118 CrossRef CAS.
- Z. Zhong, Y. Mastai, Y. Koltypin, Y. Zhao and A. Gedanken, Chem. Mater., 1999, 11, 2350–2359 CrossRef CAS.
- Y. J. Wang, A. D. Price and F. Caruso, J. Mater. Chem., 2009, 19, 6451–6464 RSC.
- F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef CAS.
- X. Y. Shi, M. W. Shen and H. Möhwald, Prog. Polym. Sci., 2004, 29, 987–1019 CrossRef CAS.
- F. Caruso, M. Spasova, A. Susha, M. Giersig and R. A. Caruso, Chem. Mater., 2000, 13, 109–116 CrossRef.
- C. J. Martinez, B. Hockey, C. B. Montgomery and S. Semancik, Langmuir, 2005, 21, 7937–7944 CrossRef CAS.
- A. A. Antipov, G. B. Sukhorukov, Y. A. Fedutik, J. Hartmann, M. Giersig and H. Möhwald, Langmuir, 2002, 18, 6687–6693 CrossRef CAS.
- S. Kidambi, J. Dai, J. Li and M. L. Bruening, J. Am. Chem. Soc., 2004, 126, 2658–2659 CrossRef CAS.
- C. G. Tian, B. D. Mao, E. B. Wang, Z. H. Kang, Y. L. Song, C. L. Wang and S. H. Li, J. Phys. Chem. C, 2007, 111, 3651–3657 CAS.
- F. Caruso, X. Shi, R. A. Caruso and A. Susha, Adv. Mater., 2001, 13, 740–744 CrossRef CAS.
- D. Wang and F. Caruso, Chem. Mater., 2002, 14, 1909–1913 CrossRef CAS.
- V. Valtchev, Chem. Mater., 2002, 14, 956–958 CrossRef CAS.
- M. Agrawal, S. Gupta and M. Stamm, J. Mater. Chem., 2011, 21, 615–627 RSC.
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., Int. Ed., 2006, 45, 813–816 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- Y. Mei, G. Sharma, Y. Lu, M. Ballauff, M. Drechsler, T. Irrgang and R. Kempe, Langmuir, 2005, 21, 12229–12234 CrossRef CAS.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Möller and J. Breu, Science, 2009, 323, 617–620 CrossRef CAS.
- G. Sharma and M. Ballauff, Macromol. Rapid Commun., 2004, 25, 547–552 CrossRef CAS.
- Y. Lu, J. Yuan, F. Polzer, M. Drechsler and J. Preussner, ACS Nano, 2010, 4, 7078–7086 CrossRef CAS.
- C.-W. Chen, M.-Q. Chen and M. Akashi, Chem. Commun., 1998, 831–832 RSC.
- C.-W. Chen, M.-Q. Chen, T. Serizawa and M. Akashi, Adv. Mater., 1998, 10, 1122–1126 CrossRef CAS.
- A. Dokoutchaev, J. T. James, S. C. Koene, S. Pathak, G. K. S. Prakash and M. E. Thompson, Chem. Mater., 1999, 11, 2389–2399 CrossRef CAS.
- M. Schrinner, F. Polzer, Y. Mei, Y. Lu, B. Haupt, M. Ballauff, A. Goldel, M. Drechsler, J. Preussner and U. Glatzel, Macromol. Chem. Phys., 2007, 208, 1542–1547 CrossRef CAS.
- H. Tamai and H. Yasuda, J. Colloid Interface Sci., 1999, 212, 585–588 CrossRef CAS.
- A. Ethirajan and K. Landfester, Chem.–Eur. J., 2010, 16, 9398–9412 CrossRef CAS.
- A. Ethirajan, U. Ziener and K. Landfester, Chem. Mater., 2009, 21, 2218–2225 CrossRef CAS.
- S. Schachschal, A. Pich and H. J. Adler, Colloid Polym. Sci., 2007, 285, 1175–1180 CAS.
- K. Schöller, A. Ethirajan, A. Zeller and K. Landfester, Macromol. Chem. Phys., 2011, 212, 1165–1175 CrossRef.
- R. Sauer, P. Froimowicz, K. Schöller, J. M. Cramer, S. Ritz, V. Mailänder and K. Landfester, Chem.–Eur. J., 2012, 18, 5201–5212 CrossRef CAS.
- V. Fischer, I. Lieberwirth, G. Jakob, K. Landfester and R. Muñoz-Espí, Adv. Funct. Mater., 2013, 23, 451–466 CrossRef CAS.
- M. Agrawal, A. Pich, N. E. Zafeiropoulos, S. Gupta, J. Pionteck, F. Simon and M. Stamm, Chem. Mater., 2007, 19, 1845–1852 CrossRef CAS.
- M. Agrawal, S. Gupta, A. Pich, N. E. Zafeiropoulos and M. Stamm, Chem. Mater., 2009, 21, 5343–5348 CrossRef CAS.
- A. Pich, J. Hain, Y. Prots and H. J. Adler, Polymer, 2005, 46, 7931–7944 CrossRef CAS.
- M. Agrawal, A. Pich, N. E. Zafeiropoulos and M. Stamm, Colloid Polym. Sci., 2008, 286, 593–601 CAS.
- M. Agrawal, A. Pich, S. Gupta, N. E. Zafeiropoulos, P. Simon and M. Stamm, Langmuir, 2008, 24, 1013–1018 CrossRef CAS.
- Y. Lu, M. Hoffmann, R. S. Yelamanchili, A. Terrenoire, M. Schrinner, M. Drechsler, M. W. Möller, J. Breu and M. Ballauff, Macromol. Chem. Phys., 2009, 210, 377–386 CrossRef CAS.
- M. Agrawal, A. Pich, S. Gupta, N. E. Zafeiropoulos, P. Formanek, D. Jehnichen and M. Stamm, Langmuir, 2010, 26, 526–532 CrossRef CAS.
- M. Das, H. Zhang and E. Kumacheva, Annu. Rev. Mater. Res., 2006, 117–142 CrossRef CAS.
- M. Karg and T. Hellweg, Curr. Opin. Colloid Interface Sci., 2009, 14, 438–450 CrossRef CAS.
- J. Texter, Colloid Polym. Sci., 2009, 287, 313–321 CAS.
- M. Antonietti, F. Gröhn, J. Hartmann and L. Bronstein, Angew. Chem., Int. Ed. Engl., 1997, 36, 2080–2083 CrossRef CAS.
- S. Xu, J. Zhang, C. Paquet, Y. Lin and E. Kumacheva, Adv. Funct. Mater., 2003, 13, 468–472 CrossRef CAS.
- M. Kuang, D. Y. Wang, M. Y. Gao, J. Hartmarm and H. Möhwald, Chem. Mater., 2005, 17, 656–660 CrossRef CAS.
- A. Sugawara, S. Yamane and K. Akiyoshi, Macromol. Rapid Commun., 2006, 27, 441–446 CrossRef CAS.
- S. Schachschal, A. Pich and H. J. Adler, Langmuir, 2008, 24, 5129–5134 CrossRef CAS.
- S. Yamane, A. Sugawara, A. Watanabe and K. Akiyoshi, J. Bioact. Compat. Polym., 2009, 24, 151–168 CrossRef CAS.
- H. B. Zhu, Y. X. Li, R. Q. Qiu, L. Shi, W. T. Wu and S. Q. Zhou, Biomaterials, 2012, 33, 3058–3069 CrossRef CAS.
- M. P. Pileni, Adv. Colloid Interface Sci., 1993, 46, 139–163 CrossRef CAS.
- M. P. Pileni, Langmuir, 1997, 13, 3266–3276 CrossRef CAS.
- M.-P. Pileni, Nat. Mater., 2003, 2, 145–150 CrossRef CAS.
- D. J. McClements, Adv. Colloid Interface Sci., 2012, 174, 1–30 CrossRef CAS.
- C. Solans, P. Izquierdo, J. Nolla, N. Azemar and M. J. Garcia-Celma, Curr. Opin. Colloid Interface Sci., 2005, 10, 102–110 CrossRef CAS.
- C. Solans and I. Solé, Curr. Opin. Colloid Interface Sci., 2012, 17, 246–254 CrossRef CAS.
- J. Bao, Y. Liang, Z. Xu and L. Si, Adv. Mater., 2003, 15, 1832–1835 CrossRef CAS.
- H. Wang, Y. Song, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2006, 128, 9284–9285 CrossRef CAS.
- H. Chen, J. He, C. Zhang and H. He, J. Phys. Chem. C, 2007, 111, 18033–18038 CAS.
- L. Dong, Y. Chu, Y. Zhang, Y. Liu and F. Yang, J. Colloid Interface Sci., 2007, 308, 258–264 CrossRef CAS.
- X. L. Yu, C. B. Cao, H. S. Zhu, Q. S. Li, C. L. Liu and Q. H. Gong, Adv. Funct. Mater., 2007, 17, 1397–1401 CrossRef CAS.
- J. Huang, Y. Xie, B. Li, Y. Liu, Y. Qian and S. Zhang, Adv. Mater., 2000, 12, 808–811 CrossRef CAS.
- J. Eastoe, M. J. Hollamby and L. Hudson, Adv. Colloid Interface Sci., 2006, 128, 5–15 CrossRef.
- M. Boutonnet, S. Lögdberg and E. Elm Svensson, Curr. Opin. Colloid Interface Sci., 2008, 13, 270–286 CrossRef CAS.
- K. Holmberg, J. Colloid Interface Sci., 2004, 274, 355–364 CrossRef CAS.
- D. H. M. Buchold and C. Feldmann, Nano Lett., 2007, 7, 3489–3492 CrossRef CAS.
- P. Leidinger, R. Popescu, D. Gerthsen and C. Feldmann, Small, 2010, 6, 1886–1891 CrossRef CAS.
- F. Gyger, M. Hubner, C. Feldmann, N. Barsan and U. Weimar, Chem. Mater., 2010, 22, 4821–4827 CrossRef CAS.
- C. Zurmühl, R. Popescu, D. Gerthsen and C. Feldmann, Solid State Sci., 2011, 13, 1505–1509 CrossRef.
- F. Bellezza, A. Cipiciani, U. Costantino, F. Marmottini and M. A. Quotadamo, Colloid Polym. Sci., 2006, 285, 19–25 CAS.
- F. Bellezza, A. Cipiciani, U. Costantino, M. Nocchetti and T. Posati, Eur. J. Inorg. Chem., 2009, 2603–2611 CrossRef CAS.
- T. Posati, F. Bellezza, A. Cipiciani, F. Costantino, M. Nocchetti, L. Tarpani and L. Latterini, Cryst. Growth Des., 2010, 10, 2847–2850 CAS.
- J. P. Ge, W. Chen, L. P. Liu and Y. D. Li, Chem.–Eur. J., 2006, 12, 6552–6558 CrossRef CAS.
- M. Sánchez-Domínguez, M. Boutonnet and C. Solans, J. Nanopart. Res., 2009, 11, 1823–1829 CrossRef.
- M. Sánchez-Domínguez, H. Koleilat, M. Boutonnet and C. Solans, J. Dispersion Sci. Technol., 2011, 32, 1765–1770 CrossRef.
- M. Sánchez-Domínguez, L. F. Liotta, G. Di Carlo, G. Pantaleo, A. M. Venezia, C. Solans and M. Boutonnet, Catal. Today, 2010, 158, 35–43 CrossRef.
- C. Okoli, M. Sánchez-Domínguez, M. Boutonnet, S. Järås, C. Civera, C. Solans and G. R. Kuttuva, Langmuir, 2012, 28, 8479–8485 CrossRef CAS.
- R. Muñoz-Espí, C. K. Weiss and K. Landfester, Curr. Opin. Colloid Interface Sci., 2012, 17, 212–224 CrossRef.
- M. Willert, R. Rothe, K. Landfester and M. Antonietti, Chem. Mater., 2001, 13, 4681–4685 CrossRef CAS.
- A. Taden, M. Antonietti, A. Heilig and K. Landfester, Chem. Mater., 2004, 16, 5081–5087 CrossRef CAS.
- R. Rossmanith, C. K. Weiss, J. Geserick, N. Hüsing, U. Hormann, U. Kaiser and K. Landfester, Chem. Mater., 2008, 20, 5768–5780 CrossRef CAS.
- N. Nabih, R. Schiller, I. Lieberwirth, E. Kockrick, R. Frind, S. Kaskel, C. K. Weiss and K. Landfester, Nanotechnology, 2011, 22, 135606 CrossRef.
- R. Schiller, C. K. Weiss and K. Landfester, Nanotechnology, 2010, 21, 405603 CrossRef.
- P. Dolcet, M. Casarin, C. Maccato, L. Bovo, G. Ischia, S. Gialanella, F. Mancin, E. Tondello and S. Gross, J. Mater. Chem., 2012, 22, 1620–1626 RSC.
- L. M. Qi, Coord. Chem. Rev., 2010, 254, 1054–1071 CrossRef CAS.
- R. H. Dong, W. M. Liu and J. C. Hao, Acc. Chem. Res., 2012, 45, 504–513 CrossRef CAS.
- H. L. Xu and W. Z. Wang, Angew. Chem., Int. Ed., 2007, 46, 1489–1492 CrossRef CAS.
- H. P. Cong and S. H. Yu, Adv. Funct. Mater., 2007, 17, 1814–1820 CrossRef CAS.
- N. Zhao and L. Qi, Adv. Mater., 2006, 18, 359–362 CrossRef CAS.
- Q. Zhao, Y. Gao, X. Bai, C. Wu and Y. Xie, Eur. J. Inorg. Chem., 2006, 2006, 1643–1648 CrossRef.
- Q. Liu, H. Liu, M. Han, J. Zhu, Y. Liang, Z. Xu and Y. Song, Adv. Mater., 2005, 17, 1995–1999 CrossRef CAS.
- G. Chen, D. Xia, Z. Nie, Z. Wang, L. Wang, L. Zhang and J. Zhang, Chem. Mater., 2007, 19, 1840–1844 CrossRef CAS.
- Y. Xu, D. Chen, X. Jiao and K. Xue, J. Phys. Chem. C, 2007, 111, 16284–16289 CAS.
- X.-H. Li, D.-H. Zhang and J.-S. Chen, J. Am. Chem. Soc., 2006, 128, 8382–8383 CrossRef CAS.
- Z. Dai, J. Zhang, J. Bao, X. Huang and X. Mo, J. Mater. Chem., 2007, 17, 1087–1093 RSC.
| This journal is © The Royal Society of Chemistry 2013 |