 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,4-Oxazine†
R. Alan
Aitken
*,
Kati M.
Aitken
,
Philip G.
Carruthers
,
Marc-Alexandre
Jean
and
Alexandra M. Z.
Slawin
EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: raa@st-and.ac.uk
First published on 23rd October 2013
Abstract
The fundamental heterocyclic compound 1,4-oxazine has been generated using FVP. It is the first parent heterocycle among all the possible isomeric oxazines, thiazines and their heavier atom analogues to be characterised spectroscopically, and is shown to exist in solution entirely as the 4H-isomer. X-ray structure determination of the N-Boc precursor shows significant deviations from theoretically predicted geometric parameters.
While six-membered ring heterocycles containing two group 15 atoms, exemplified by the three isomeric diazines, are well known, stable aromatic compounds, neutral analogues containing one or more group 16 atoms cannot be aromatic and are much less stable. For example, although both 1,4-dioxin1 and 1,4-dithiin2 were prepared at an early date, these compounds are rather unstable and highly reactive towards addition and polymerisation.1,3 Among the many possible parent six-membered ring heterocycles containing one group 15 atom and one group 16 atom, none appear to have been so far isolated or spectroscopically characterised, with the sole exception of 1,4-thiazine which was the subject of a single report in 1948,4 not subsequently reproduced.
1,4-Oxazine 1 is among the simplest heterocyclic compounds to be so far unknown. Although a fair number of substituted and ring-fused 1,4-oxazines are known, the parent compound has never been prepared and work on it has so far been restricted to theoretical predictions of bond lengths and electron distribution.5 Our long-standing interest in the area started with the unexpected formation of 1,4-dioxin in a retro-Diels–Alder reaction under conditions of flash vacuum pyrolysis (FVP), which allowed its NMR spectra to be recorded for the first time.6 However numerous attempts to extend the retro-Diels–Alder approach to formation of 1,4-oxazine by pyrolysis of derivatives 2 or 3 failed. Synthesis analogous to that of 1,4-thiazine by pyrolysis of cyclic imide 4, either with or without a reducing metal, was also unsuccessful as other processes intervened (Scheme 1).7
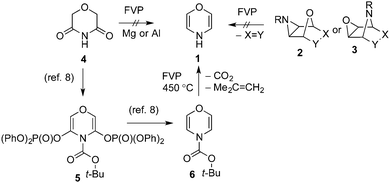 | ||
| Scheme 1 Routes to 1,4-oxazine. | ||
A breakthrough in the area occurred in 2007 when Coudert, Gillaizeau and co-workers described the three-step conversion of 4via5 into 6, thus making a simple monosubstituted 1,4-oxazine readily available for the first time.8 We now report the removal of the tert-butoxycarbonyl group from 6 to generate the parent 1,4-oxazine 1 together with its spectroscopic characterisation. Wasserman achieved thermal deprotection of an aliphatic N-tert-butoxycarbonyl amine in the course of a synthesis of the alkaloid homaline by boiling either in quinoline or diphenyl ether,9 and a short time later Cava extended this method to heterocyclic derivatives when he described removal of N-tert-butoxycarbonyl groups from pyrroles and indoles in almost quantitative yield simply by heating the solids at 180–185 °C.10
This suggested to us that FVP might be the ideal technique for conversion of 6 into 1: the purely thermal reaction involving no acids or bases would be further encouraged by high vacuum since two molecules of inert gaseous by-products are formed – isobutene and CO2. The experimental set-up would also produce the oxazine directly in the liquid nitrogen-cooled cold trap giving the best chance for its isolation. In a brief model study, both N-tert-butoxycarbonylpiperidine and -pyrrole behaved well giving excellent yields of deprotected products together with isobutene and CO2 at 450–500 °C and 10−2 Torr. When a sample of compound 6 was subjected to FVP at 450 °C and 10−2 Torr it gave some unreacted starting material at the furnace exit and in the cold trap an oil which was washed out with CDCl3 for analysis.‡ The spectrum proved to be extremely clean with peaks due to isobutene, the starting material 6 and a new A2B2 pattern at δ 5.19 and 5.29 (J 4.5 Hz) attributed to 1. The NH appeared as a broad peak centred at δ 2.92. In the 13C NMR spectrum all peaks were attributable to starting material 6, isobutene, and 1,4-oxazine 1 with signals at δ 114.5 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N) and 126.4 (CH–O). A comparison of these chemical shifts with those for comparable ring systems is shown in Fig. 1.
CH–N) and 126.4 (CH–O). A comparison of these chemical shifts with those for comparable ring systems is shown in Fig. 1.
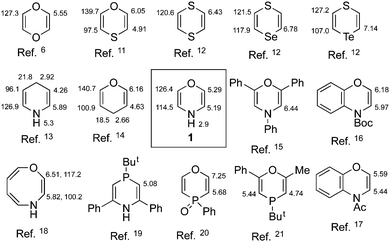 | ||
| Fig. 1 1H and 13C NMR chemical shifts of 1 compared with related heterocycles. | ||
Both C and H values are almost halfway between those for the corresponding atoms in pyran and dihydropyridine – in some sense it may be considered a hybrid of these two structures. The 1H chemical shift values are markedly lower than for the vinylogous 10π-1,4-oxazocine indicating the complete absence of aromaticity for this 8π-system. There was no evidence at all of the isomeric 2H structure which is perhaps surprising given the ready interconversion observed in such similar systems as 1H- and 3H-azepine.22 In an early report of 1H NMR data supporting the transient existence of 2H-1,4-benzoxazine, the spectrum was obtained in TFA as solvent and so clearly refers to a protonated species.23
A fully coupled 13C NMR spectrum allowed determination of C–H coupling constants for 1 as shown in Table 1 and these make an interesting comparison with the values for 6, 1,4-dioxin,6 1,4-dithiin, 1,4-thiaselenin and 1,4-thiatellurin.12
| X | Y | 1 J C2–H2 | 2 J C2–H3 | 3 J C2–H6 | 1 J C3–H3 | 2 J C3–H2 | 3 J C3–H5 | Ref. |
|---|---|---|---|---|---|---|---|---|
| a This work. b Two separate values for 6 due to restricted rotation of Boc.8 | ||||||||
| O | NH | 196.9 | 10.8 | 5.2 | 178.4 | 14.0 | 5.2 | |
| O | NBoc | 197.6 | 11.1 | 6.2 | 187.8 | 14.9 | 3.2 | , |
| 198.4 | 11.1 | 6.3 | 187.8 | 14.9 | 3.3 | |||
| O | O | 197.2 | 16.4 | ∼0 | 197.2 | 16.4 | ∼0 | 6 |
| S | S | 179.2 | 8.1 | ∼0 | 179.2 | 8.1 | ∼0 | 12 |
| S | Se | 178.8 | 7.7 | 2.0 | 181.2 | 7.7 | ∼0 | 12 |
| S | Te | 177.8 | 6.2 | 1.9 | 177.5 | 5.8 | ∼0 | 12 |
The 14N NMR spectrum was also recorded showing two signals at δ −276 (6) and −325 (1) relative to MeNO2. These are assigned on the basis that after 6 h the signal at −325 had completely disappeared, while that at −276 remained unchanged and was shown by a 2D experiment to correlate with the 1H NMR signals for 6. We were also able to obtain a HRMS measurement.
It should be mentioned that 1,4-oxazine seems to be stabilised in solution by the presence of the unreacted precursor 6, perhaps by a hydrogen bonding interaction. FVP of 6 at temperatures of 500 °C and higher led to complete conversion to give a product which decomposed in CDCl3 before spectra could be obtained. Attempts to record the spectra of the 450 °C pyrolysis products in C6D6 likewise led to immediate polymerisation of 1 and only signals for 6 and isobutene were detected.
Finally, in the course of the repeated preparations of 6 required for this work, the compound which Coudert and coworkers reported as a colourless oil was observed to sublime in a closed flask upon storage at −30 °C for several months to give colourless crystals, mp 35–36 °C, suitable for X-ray diffraction. The resulting structure (Fig. 2),24 the first for a monosubstituted 1,4-oxazine, shows a marked difference from the data predicted theoretically for 1,4-oxazine 1, both in the bond lengths where there are substantially shorter CC and C–N distances,25 and the deviation of O and N from the plane formed by the four ring carbon atoms.26 The deviation from planarity in 6 is however nowhere near so great as in the 3-methoxycarbonyl derivative 7, the only other simple 1,4-oxazine to be crystallographically characterised,27 which also has unequal bond lengths and a distinctly pyramidal nitrogen (angle sum at N: 7 355.5° vs.6 359.8°).
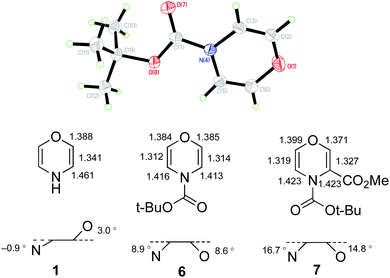 | ||
| Fig. 2 ORTEP diagram (50% probability level) for 6 and comparison of measured bond lengths and ring shape for 6 and 7 with theoretical values for 1. | ||
Although 1,4-oxazine 1 can now be generated and observed spectroscopically, it is not at all stable and decomposes in CDCl3 solution to give brown insoluble, probably polymeric, products with a half-life at RT of about one hour.
The authors thank Dr T. Lebl for running the 14N NMR spectrum.
Notes and references
- R. K. Summerbell and R. R. Umhoefer, J. Am. Chem. Soc., 1939, 61, 3020–3022 CrossRef CAS.
- W. E. Parham, H. Wynberg and F. L. Ramp, J. Am. Chem. Soc., 1953, 75, 2065–2069 CrossRef CAS.
- W. E. Parham, B. Gadsby and R. A. Mikulec, J. Org. Chem., 1959, 24, 1819–1822 CrossRef CAS.
- C. Barkenbus and P. S. Landis, J. Am. Chem. Soc., 1948, 70, 684–685 CrossRef CAS.
- R. A. Aitken and K. M. Aitken, in Compr. Heterocycl. Chem. III, ed. R. A. Aitken, Elsevier, Oxford, 2008, vol. 8, pp. 461–511 Search PubMed.
- R. A. Aitken, J. I. G. Cadogan and I. Gosney, J. Chem. Soc., Perkin Trans. 1, 1994, 927–931 RSC.
- R. A. Aitken, D. M. M. Farrell and E. H. M. Kirton, Chem. Heterocycl. Compd. (Engl. Transl.), 2001, 37, 1526–1531 CrossRef CAS.
- E. Claveau, I. Gillaizeau, J. Blu, A. Bruel and G. Coudert, J. Org. Chem., 2007, 72, 4832–4836 CrossRef CAS PubMed.
- H. H. Wasserman, G. D. Berger and K. R. Cho, Tetrahedron Lett., 1982, 23, 465–468 CrossRef CAS; H. H. Wasserman and G. D. Berger, Tetrahedron, 1983, 39, 2459–2464 CrossRef.
- V. H. Rawal and M. P. Cava, Tetrahedron Lett., 1985, 26, 6141–6142 CrossRef CAS; V. H. Rawal, R. J. Jones and M. P. Cava, J. Org. Chem., 1987, 52, 19–28 CrossRef.
- M. Schoufs, J. Meijer and L. Brandsma, Recl. Trav. Chim. Pays-Bas, 1980, 99, 12–14 CrossRef CAS.
- J. Meijer, P. Vermeer, H. D. Verkruijsse and L. Brandsma, Recl. Trav. Chim. Pays-Bas, 1973, 92, 1326–1330 CrossRef CAS.
- A. J. de Koning, P. H. M. Budzelaar, J. Boersma and G. J. M. van der Kerk, J. Organomet. Chem., 1980, 199, 153–169 CrossRef CAS.
- B. Engels, J. C. Schöneboom, A. F. Münster, S. Groetsch and M. Christl, J. Am. Chem. Soc., 2002, 124, 287–297 CrossRef CAS PubMed.
- J. Correia, J. Org. Chem., 1973, 38, 3433–3434 CrossRef CAS.
- C. Buon, L. Chacun-Lefèvre, R. Rabot, P. Bouyssou and G. Coudert, Tetrahedron, 2000, 56, 605–614 CrossRef CAS.
- H. Bartsch, W. Kropp and M. Pailer, Monatsh. Chem., 1979, 110, 267–278 CrossRef CAS.
- B. Zipperer, D. Hunkler, H. Fritz, G. Rihs and H. Prinzbach, Angew. Chem., Int. Ed. Engl., 1984, 23, 309–311 CrossRef.
- G. Märkl and D. Matthes, Angew. Chem., Int. Ed. Engl., 1972, 11, 1019–1020 CrossRef.
- M. Maumy, Bull. Soc. Chim. Fr., 1972, 1600–1603 CAS.
- G. Märkl, G. Adolin, F. Kees and G. Zander, Tetrahedron Lett., 1977, 18, 3445–3448 CrossRef.
- E. Vogel, H.-J. Altenbach, J.-M. Drossard, H. Schmickler and H. Stegelmeier, Angew. Chem., Int. Ed. Engl., 1980, 19, 1016–1018 CrossRef.
- F. Chioccara, G. Prota and R. H. Thomson, Tetrahedron, 1976, 32, 1407–1409 CrossRef CAS.
- CCDC 956939.
- N. Trinajstic, J. Mol. Struct., 1971, 8, 236–239 CrossRef CAS.
- O. Y. Borbulevych and O. V. Shishkin, J. Mol. Struct., 1998, 446, 11–14 CrossRef CAS.
- E. Claveau, I. Gillaizeau, J. Kalinowska-Tluscik, P. Bouyssou and G. Coudert, J. Org. Chem., 2009, 74, 2911–2914 CrossRef CAS PubMed.
Footnotes |
| † CCDC 956939. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc47801g |
| ‡ Experimental procedure for generation of 4H-1,4-oxazine 1: a sample of 6 (ref. 8) (0.2 g) was volatilised through a 30 × 2.5 cm fused quartz tube heated within a horizontal laboratory tube furnace at 450 °C and maintained at a pressure of 10−2 Torr by a rotary oil pump. The contact time in the hot zone under these conditions was estimated to be ca. 10 ms. The products were collected in a liquid nitrogen cooled U-shaped trap and, after the pyrolysis was complete, were dissolved out in CDCl3 for analysis. The spectra showed the presence of unchanged 6, isobutene, and 1: δH (300 MHz, CDCl3) 5.29 and 5.19 (4H, A2B2 pattern, 3J(H–H) 4.5 Hz) and 2.92 (1H, br s); δC (75 MHz, CDCl3) 126.4 (CH–O) and 114.5 (CH–N); δC (undecoupled) 126.4 (ddd, J 196.9, 10.8 and 5.2 Hz) and 114.5 (ddd, J 178.4, 14.0 and 5.2 Hz); HRMS (ES−): found 84.0462. C4H6NO (MH−) requires 84.0449. |
| This journal is © The Royal Society of Chemistry 2013 |