 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Epoxidation of bromoallenes connects red algae metabolites by an intersecting bromoallene oxide – Favorskii manifold†
D.
Christopher Braddock
*,
James
Clarke
and
Henry S.
Rzepa
Department of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: c.braddock@imperial.ac.uk; Fax: +44 (0)2075945805; Tel: +44 (0)2075945772
First published on 23rd October 2013
Abstract
DMDO epoxidation of bromoallenes gives directly α,β-unsaturated carboxylic acids under the reaction conditions. Calculated (ωB97XD/6-311G(d,p)/SCRF = acetone) potential energy surfaces and 2H- and 13C-labeling experiments are consistent with bromoallene oxide intermediates which spontaneously rearrange via a bromocyclopropanone in an intersecting bromoallene oxide – Favorskii manifold.
The remarkably wide structural diversity and complexity of halogenated C15 acetogenin metabolites isolated from marine red algae of Laurencia species1 continue to stimulate innovative efforts in their target synthesis,2 in the discovery of new synthetic transformations3 and in advancing biosynthetic hypotheses.4 A recent re-isolation5 of obtusallene IV (1)6 from Laurencia marilzae provided also 12-epoxyobtusallene IV (2) and unnamed α,β-unsaturated carboxylate ester (3) with an identical macrocycle to epoxybromoallene 2 (Fig. 1). It seems reasonable to connect E-alkene 1 and trans-epoxide 2 biogenetically via enzymatic epoxidation,7 and on the basis of their co-isolation, we propose that bromoallene 2 and α,β-unsaturated carboxylate 3 may also be connected biogenetically by epoxidation.
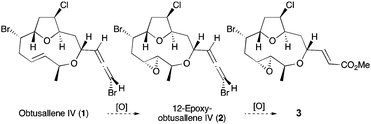 | ||
| Fig. 1 Metabolites 1–3 from Laurencia marilzae and proposed biogenesis via epoxidation events. | ||
While the epoxidation of allenes8,9 and vinyl bromides10 has been studied, the epoxidation of bromoallenes has not been reported.11 Herein, we report the hitherto unknown direct conversion of bromoallenes to α,β-unsaturated carboxylic acids via an initial epoxidation event and the presumed intermediacy of a bromoallene oxide. We also show by computational modeling and 2H- and 13C-labeling studies that the latter's spontaneous reorganization to an α,β-unsaturated carboxylic acid under the reaction conditions is consistent with a bromocyclopropanone intermediate in an intersecting allene oxide – Favorskii manifold.
Bromoallene 412 was selected as a suitable substrate for investigating epoxidation and was synthesized by a standard sequence from heptanal (ESI†).13 Much to our delight, epoxidation of bromoallene 4 using dimethyl dioxirane (DMDO), generated either in situ14 or as a solution (ESI†)15 (Scheme 1), gave a mixture of Z and E-α,β-unsaturated carboxylic acids 5 directly in low but reproducible yields (note §, ESI†). The low yields can be attributed to decomposition of DMDO16a under the reaction conditions to methyl radicals,16b and subsequent radical attack on either of the products or starting materials (note ¶, ESI†).
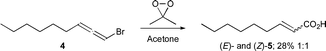 | ||
| Scheme 1 Epoxidation of bromoallene 4 using DMDO solution. | ||
Mechanistically, we invoke the following pathway for the formation of α,β-unsaturated carboxylic acids from DMDO mediated epoxidation of bromoallenes (Fig. 2). Initial epoxidation of the bromoallene would give bromoallene oxides of the type A and/or B (note ¥, ESI†). Spontaneous epoxide opening8cvia bromo oxyallyl cations C and D (note ††, ESI†) respectively converge on the same bromocyclopropanone E. This intermediate now intersects with the Favorskii rearrangement manifold of α,α- and α,α′-dibromoketones where the resulting bromocyclopropanones E are known to collapse after attack by water giving hydrate F to α,β-unsaturated carboxylic acids 5 (note **, ESI†).17,18 Evidently, there is sufficient water in the dioxirane solution to function as a nucleophile here (note ‡‡, ESI†). Interestingly, regardless of the initial site of epoxidation, this mechanism predicts that carbon atoms 1 and 2 in bromoallene 4 interchange positions in the α,β-unsaturated carboxylic acid products 5.
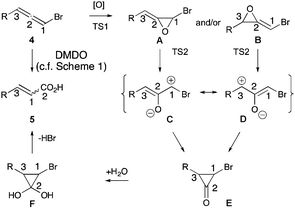 | ||
| Fig. 2 Mechanistic rationale for conversion of bromoallenes into α,β-unsaturated carboxylic acids, with the carbon atoms of the functional groups numbered 1–3 showing an interchange of carbon atoms 1 and 2 (see also interactive Fig. 2 in HTML version of this article). | ||
This mechanism can be subjected to scrutiny via density functional level (ωB97XD/6-311G(d,p)/SCRF = acetone)19 exploration of the potential energy surface (R = H, Me, presented as an interactive version of Fig. 2 (ref. 20) via a digital data repository21). Oxygen transfer from dimethyldioxirane to form both A and B (TS1) have thermally accessible free energy activation barriers 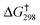 (R = H, 26.8 for A, 27.3 for B; R = Me, 26.8 for A, 24.6 kcal mol−1 for B), followed by a second, lower energy dyotropic rearrangement (TS2) to give E. An intrinsic reaction coordinate (IRC) reveals that TS2 (R = H,Me) represents the concerted transformation of A or B to E, with C/D acting as “hidden intermediates” in the process.22 Such hidden intermediates can be potentially transformed to real ones by tuning the substituents, and in this instance changing R from H or Me to OMe is predicted to accomplish this by stabilization of C/D (see interactive Fig. 2). TS2 itself (R = Me) has some early character of C/D; the C–Br bond is calculated to initially contract in length due to a significant stabilising resonance contribution of Br lone pairs, from 1.924/1.896 Å (A and B respectively) via 1.840/1.885 (TS2), 1.856/1.868 (C/D acting as hidden intermediates) to 1.921/1.922 Å (E).23 Calculations having demonstrated the thermal accessibility of the epoxidation-bromocyclopropanone sequence, 2H- and 13C-labeling experiments were necessary to verify the overall reorganization (4 to A/B to E to F to 5, Fig. 2) of the carbon framework.24
(R = H, 26.8 for A, 27.3 for B; R = Me, 26.8 for A, 24.6 kcal mol−1 for B), followed by a second, lower energy dyotropic rearrangement (TS2) to give E. An intrinsic reaction coordinate (IRC) reveals that TS2 (R = H,Me) represents the concerted transformation of A or B to E, with C/D acting as “hidden intermediates” in the process.22 Such hidden intermediates can be potentially transformed to real ones by tuning the substituents, and in this instance changing R from H or Me to OMe is predicted to accomplish this by stabilization of C/D (see interactive Fig. 2). TS2 itself (R = Me) has some early character of C/D; the C–Br bond is calculated to initially contract in length due to a significant stabilising resonance contribution of Br lone pairs, from 1.924/1.896 Å (A and B respectively) via 1.840/1.885 (TS2), 1.856/1.868 (C/D acting as hidden intermediates) to 1.921/1.922 Å (E).23 Calculations having demonstrated the thermal accessibility of the epoxidation-bromocyclopropanone sequence, 2H- and 13C-labeling experiments were necessary to verify the overall reorganization (4 to A/B to E to F to 5, Fig. 2) of the carbon framework.24
Deuterated bromoallene (1-2H)-4 was prepared by addition of ethynylmagnesium bromide to heptanal, in situ deprotonation of the propargylic alkoxide with n-butyllithium and quenching with MeOH-d4 to give labeled propargylic alcohol (1-2H)-6 (Scheme 2). Subsequent alcohol trisylation25 gave (1-2H)-7, and SN2′ displacement of the trisylate with bromide under the action of LiCuBr2 (ref. 26) provided bromoallene (1-2H)-4 with 70% deuterium incorporation at the 1-position.†
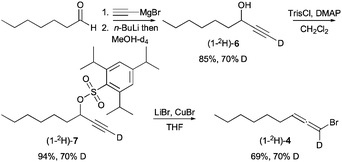 | ||
| Scheme 2 Synthesis of deuterated bromoallene (1-2H)-4. | ||
13C-labeled bromoallene (1-13C)-4 was similarly targeted, commencing with silyl enol ether 8 formation27 from octanal (Scheme 3). Oxidation using mCPBA gave interrupted Rubottom28 adduct 9, which could be acetylated to give acetate 10. Desilylation using buffered TBAF29 revealed protected α-hydroxyaldehyde 11, which we planned to use in a Wittig reaction with a suitably 13C-labeled phosphorous ylid. To the best of our knowledge, there is only a single report30 using methyltriphenylphosphonium iodide to generate the Stork–Wittig reagent31 using an in situ deprotonation–iodination–deprotonation procedure which we adapted using 13C-labeled salt 12 – available from relatively inexpensive 99% atom 13C-labeled methyl iodide – to give vinyl iodides Z-(1-13C)-13, E-(1-13C)-13 and diiodide (1-13C)-14.32 Acetate deprotection as a mixture gave the corresponding alcohols Z-(1-13C)-15, E-(1-13C)-15 and (1-13C)-16 all with 99% 13C at the alkene terminus.†
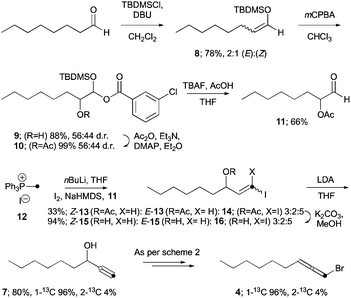 | ||
| Scheme 3 Synthesis of 13C-labeled bromoallene (1-13C)-4. | ||
Dehydrohalogenation of Z- and E-iodides (1-13C)-15 in the presence of inseparable diiodide (1-13C)-16 with LDA gave propargylic alcohol (1-13C)-7 in good overall yield, with the unprecedented observation that LDA converts vinyl 1,1-diiodides into terminal alkynes also (note §§, ESI†). Interestingly, 4% of the alkyne product was found to be the 2-13C isotopomer (ESI†), implicating a 1,1-elimination reaction pathway for diiodide 16 and competitive alkyl group migration from a vinylidene intermediate (note ¶¶, ESI†). Alcohol (1-13C)-7 was then converted to the desired bromoallene (1-13C)-7 (as 4% of its 2-13C-isotopomer, ESI†) as previously described (cf., Scheme 2).
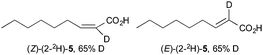
With (1-2H)-4 and (1-13C)-4 in hand, epoxidation with DMDO was conducted. For deuterated (1-2H)-4, after the reaction was conducted in the usual manner (cf., Scheme 1), E-(2-2H)-5 and Z-(2-2H)-5 were isolated each showing 65% deuteration at the α-position only (note ‡, ¥¥, ESI†). Evidently, this result is consistent with the proposed mechanism (cf., Fig. 2) (note †††, ESI†). More compellingly, epoxidation of bromoallene (1-13C)-4 gave (E-2-13C)-533 and (Z-2-13C)-5 (28% isolated yield) where carbon atoms 1 and 2 from the bromoallene have entirely interchanged positions, giving also 4% of each of the (E-1-13C)-5 and (Z-1-13C)-5 isotopomers (ESI†). The expected 1JCH coupling constants experienced by the α-vinyl protons of the major isotopomers are clearly apparent in their 1H NMR spectra (Fig. 3).
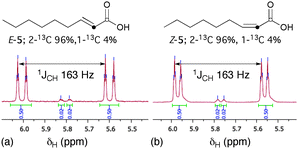 | ||
| Fig. 3 1H NMR spectra of (a) (E-2-13C)-5 and (b) (Z-2-13C)-5 displaying the expected 1JCH values for the α-vinyl protons. | ||
In conclusion we have established that the hitherto unknown direct conversion of bromoallenes to α,β-unsaturated carboxylic acids using DMDO is consistent with an initial epoxidation event (note ***, ESI†) followed by a spontaneous reorganization via a bromocyclopropanone, a mechanism supported by calculations, in an intersecting bromoallene oxide – Favorskii manifold. These experiments support the proposed biogenesis of α,β-unsaturated carboxylate 3 from bromoallene 2 by epoxidation (note ‡‡‡, ESI†).
We thank the EPSRC for DTG funding (to J. C.).
Notes and references
- (a) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munroa and M. R. Prinsep, Nat. Prod. Rep., 2013, 30, 237–323 RSC and earlier reviews in this series; (b) B.-G. Wang, J. B. Gloer, N.-Y. Ji and J.-C. Zhao, Chem. Rev., 2013, 113, 3632–3685 CrossRef CAS PubMed.
- For a comprehensive review of the synthesis of medium ring ethers from Laurencia sp., see: (a) K. Fujiwara, Top. Heterocycl. Chem., 2006, 5, 97–148 CAS; (b) For recent leading examples see: B. S. Dyson, J. W. Burton, T.-i. Sohn, B. Kim, H. Bae and D. Kim, J. Am. Chem. Soc., 2012, 134, 11781–11790 CrossRef CAS PubMed; (c) M. J. Kim, T.-i. Sohn, D. Kim and R. S. Paton, J. Am. Chem. Soc., 2012, 134, 20178–20188 CrossRef CAS PubMed and references cited therein.
- For recent representative examples see: (a) S. Keshipeddy, I. Martínez, B. F. Castillo II, M. D. Morton and A. R. Howell, J. Org. Chem., 2012, 77, 7883–7890 CrossRef CAS PubMed; (b) S. A. Snyder, A. P. Brucks, D. S. Treitler and I. Moga, J. Am. Chem. Soc., 2012, 134, 17714–17721 CrossRef CAS PubMed; (c) S. A. Snyder, D. S. Treitler, A. P. Brucks and W. Sattler, J. Am. Chem. Soc., 2011, 133, 15898–15901 CrossRef CAS PubMed; (d) N. Ortega, V. S. Martín and T. Martín, J. Org. Chem., 2010, 75, 6660–6672 CrossRef CAS PubMed and references cited therein.
- For a review see: (a) A. Murai, in Comprehensive Natural Products Chemistry, ed. D. H. R. Barton, O. Meth-Cohn and K. Nakinishi, Elsevier, Oxford, 1999, vol. 1, pp. 303–324 Search PubMed. For representative examples see: (b) K. J. Bonney and D. C. Braddock, J. Org. Chem., 2012, 77, 9574–9584 CrossRef CAS PubMed; (c) A. Gutiérrez-Cepeda, J. J. Fernández, M. Norte and M. L. Souto, Org. Lett., 2011, 13, 2690–2693 CrossRef PubMed; (d) D. C. Braddock, D. S. Millan, Y. Perez-Fuertes, R. H. Pouwer, R. N. Sheppard, S. Solanki and A. J. P. White, J. Org. Chem., 2009, 74, 1835–1841 CrossRef CAS PubMed; (e) D. C. Braddock, Org. Lett., 2006, 8, 6055–6058 CrossRef CAS PubMed and references cited therein.
- A. Gutiérrez-Cepeda, J. J. Fernández, L. V. Gil, M. López-Rodríguez, M. Norte and M. L. Souto, J. Nat. Prod., 2011, 74, 441–448 CrossRef PubMed.
- (a) G. Guella, G. Chiasera, I. Mancini, A. Öztunç and F. Pietra, Chem.–Eur. J., 1997, 3, 1223–1231 CrossRef CAS; (b) M. L. Ciavatta, M. Gavagnin, R. Puliti, G. Cimino, E. Martínez, J. Ortea and C. A. Mattia, Tetrahedron, 1997, 53, 17343–17350 CrossRef CAS.
- For reviews on enzymatic epoxidation of alkenes see: (a) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS PubMed; (b) P. R. Ortiz de Montellano and J. J. De Voss, Nat. Prod. Rep., 2002, 19, 477–493 RSC.
- For reviews see: (a) G. L'abbé, Angew. Chem., Int. Ed. Engl., 1980, 19, 276–289 CrossRef; (b) W. Smadja, Chem. Rev., 1983, 83, 263–320 CrossRef CAS; (c) T. H. Chan and B. S. Ong, Tetrahedron, 1980, 36, 2269–2289 CrossRef CAS.
- For the first isolated allene oxide and its thermal rearrangement to a cyclopropanone see: R. L. Camp and F. D. Greene, J. Am. Chem. Soc., 1968, 90, 7349 CrossRef CAS.
- For an early report on their synthesis and reactivity of 2-bromooxiranes see: (a) A. Hassner and P. Catsoulacos, J. Org. Chem., 1967, 32, 549–553 CrossRef CAS PubMed; For a representative naturally occurring 2-bromooxirane see: (b) K. Watanabe, M. Sekine and K. Iguchi, J. Nat. Prod., 2003, 66, 1434–1440 CrossRef CAS PubMed.
- There is a single report of a bromoallene oxide functionality: ethyl 2-bromo-3-(diphenylmethylene) oxirane-2-carboxylate was reported in a study of ketenes and aliphatic diazo compounds: H. Staudinger and T. Reber, Helv. Chim. Acta, 1921, 4, 3–23 CrossRef CAS.
- P. C. Ravikumar, L. Yao and F. F. Fleming, J. Org. Chem., 2009, 74, 7294–7299 CrossRef CAS PubMed.
- See for example: D. C. Braddock, R. Bhuva, Y. Pérez-Fuertes, R. Pouwer, C. A. Roberts, A. Ruggiero, E. S. E. Stokes and A. J. P. White, Chem. Commun., 2008, 1419–1421 RSC and references cited therein.
- J. K. Crandall, D. J. Batal, F. Lin, T. Reix, G. S. Nadol and R. A. Ng, Tetrahedron, 1992, 48, 1427–1448 CrossRef CAS and references therein.
- (a) R. W. Murray and M. Singh, Org. Synth., 1997, 74, 91 CrossRef CAS . For the use of cyclic ketones as dioxirane precursors see: ; (b) R. W. Murray, M. Singh and R. Jeyaraman, J. Am. Chem. Soc., 1992, 114, 1346–1351 CrossRef CAS.
- For leading examples see: (a) M. Singh and R. W. Murray, J. Org. Chem., 1992, 57, 4263–4270 CrossRef CAS; (b) N. N. Kabal'nova, D. V. Kazakov, N. M. Shishlov and V. V. Shereshovets, Russ. Chem. Bull., 1996, 45, 1481–1483 CrossRef.
- For a review of the Favorskii reaction see: J. Mann, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and G. Pattenden, Pergamon Press, Oxford, 1991, vol. 3, ch. 3.7, pp. 839–859 Search PubMed.
- In a previous isolation from red algae Bonnemaisonia, both 1,1,3-tribromo-2-ketones and their proposed Favorskii products – E- and Z-3-bromo-2-alkenoic acids – were co-isolates: O. J. McConnell and W. Fenical, Phytochemistry, 1980, 19, 233–247 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 6615–6620 RSC.
- D. C. Braddock, J. Clarke and H. S. Rzepa, Figshare, 2013, DOI:10.6084/m9.figshare.785756 and the further digital repository links therein.
- J. Downing, P. Murray-Rust, A. P. Tonge, P. Morgan, H. S. Rzepa, F. Cotterill, N. Day and M. J. Harvey, J. Chem. Inf. Model., 2008, 48, 1571–1581 CrossRef CAS PubMed . See also http://www.force11.org/AmsterdamManifesto for the Amsterdam Manifesto on data citation principles.
- D. Cremer and E. Kraka, Acc. Chem. Res., 2010, 43, 591–601 CrossRef CAS PubMed; H. S. Rzepa and C. Wentrup, J. Org. Chem., 2013, 78, 7565–7574 CrossRef PubMed.
- Similar non-linear behavior of a bond is found in the related dyotropic rearrangement of dibromoethanes; D. C. Braddock, D. Roy, D. Lenoir, E. Moore, H. S. Rzepa, J. I.-C. Wu and P. von R. Schleyer, Chem. Commun., 2012, 48, 8943–8945 RSC.
- For pioneering labeling work to elucidate the mechanism of the Favorskii rearrangement and to implicate a symmetrical intermediate, viz., a cyclopropanone see: R. B. Loftfield, J. Am. Chem. Soc., 1951, 73, 4707–4714 CrossRef CAS.
- T. A. Grese, K. D. Hutchinson and L. E. Overman, J. Org. Chem., 1993, 58, 2468–2477 CrossRef CAS.
- C. J. Elsevier, P. Vermeer, A. Gedanken and W. Runge, J. Org. Chem., 1985, 50, 364–367 CrossRef CAS.
- Y. Tanigichi, J. Inanaga and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1981, 54, 3229–3230 CrossRef.
- A. Hassner, R. H. Reuss and H. W. Pinnick, J. Org. Chem., 1975, 40, 3427–3429 CrossRef CAS.
- For a representative example see: J. S. Debenham, R. Rodebaugh and B. Fraser-Reid, J. Org. Chem., 1997, 62, 4591–4600 CrossRef CAS.
- W. Zhu, M. Jiménez, W.-H. Jung, D. P. Camarco, R. Balachandran, A. Vogt, B. W. Day and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 9175–9187 CrossRef CAS PubMed.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 2173–2174 CrossRef CAS.
- For the unwanted formation of a vinyl 1,1-diiodide in a Stork–Wittig reaction using [Ph3PCH2I]I see: P. Li, J. Li, F. Arikan, W. Ahlbrecht, M. Dieckmann and D. Menche, J. Org. Chem., 2010, 75, 2429–2444 CrossRef CAS PubMed.
- D. C. Braddock, J. Clarke and H. S. Rzepa, Figshare, 2013, DOI:10.6084/m9.figshare.785753viz. ref. 20 and 21 for explanation.
Footnote |
| † Electronic supplementary information (ESI) available: Notes §, ¶, ¥, ††, **, ‡‡, §§, ¶¶, ¥¥, †††, ***, ‡‡‡; general experimental; experimental details and characterising data for compounds leading to bromoallenes 4 (including ESI Scheme S1 for the synthesis of bromoallenes 4), (1-2H)-4 and (1-13C-4) and epoxidation thereof leading to E- and Z-5, (E-2-2H)- and (Z-2-2H)-5, and (E-2-13C)- and (Z-2-13C)-5; Copies of 1H and 13C spectra for all compounds showing 2H and 13C isotopic shifts and coupling constants where appropriate; ESI references. See DOI: 10.1039/c3cc46720a |
| This journal is © The Royal Society of Chemistry 2013 |