 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
WC@meso-Pt core–shell nanostructures for fuel cells†
Zhao-Yang
Chen
a,
Chun-An
Ma
*ab,
You-Qun
Chu
a,
Jia-Mei
Jin
b,
Xiao
Lin
b,
Christopher
Hardacre
b and
Wen-Feng
Lin
*b
aState Key Laboratory Breeding Base for Green Chemistry Synthesis Technology, International Sci. & Tech. Cooperation Base of Energy Materials and Application, College of Chemical Engineering and Materials Science, Zhejiang University of Technology, Hangzhou 310032, PR China. E-mail: science@zjut.edu.cn; Fax: +86-0571-88320830; Tel: +86-0571-88320830
bCentre for the Theory and Application of Catalysis (CenTACat), School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast BT9 5AG, UK. E-mail: w.lin@qub.ac.uk
First published on 17th October 2013
Abstract
We developed a facile method to synthesize core–shell WC@meso-Pt nanocatalysts by carburizing ammonium tungstate and copper nitrate via gas–solid reactions, followed by a Pt replacement reaction. The mesoporous nanocomposite displays higher activity and stability towards methanol electrooxidation than commercial Pt/C catalysts.
Catalysts are often structure sensitive materials which, in most cases, require complex synthesis routes. There are a wide range of synthesis techniques (e.g., gas–solid reactions, wet-chemical reactions or solid-state processes) available for constructing different kinds of nanoporous, mesoporous or core–shell structures as more effective catalysts.1–5 The initial report on core–shell systems in 1992 has been followed by intensive experimental and theoretical research.2 Hydrolysis and sol–gel are popular methods for the synthesis of core–shell materials (such as Au/SiO2, Fe3O4/SiO2, etc.).3 Gas–solid reaction, which can be an effective route for large-scale material production, has recently been explored for synthesising core–shell oxide materials.5
Tungsten carbide (WC) is an excellent candidate component for core–shell materials owing to its unusual properties, such as high melting point, superior hardness, high oxidation resistance, and superior electrical conductivity.6 WC was also shown to have a platinum-like behaviour towards the chemisorption of hydrogen and oxygen, and its applicability as an alternative electrocatalyst to Pt has been demonstrated.7 WC may enhance both the physical and chemical properties of core–shell structures8 and the coactivity of WC and Pt has been revealed recently.9 Although it is expected that WC–Pt nanocomposites with uniform core–shell structure are likely to have excellent catalytic properties,10 significant synthetic challenges remain because the unique properties of WC impede the formation of the nanoporous core–shell construction. Nevertheless, we have recently demonstrated that nanosized and mesoporous WC can be prepared by carefully designing precursors to have the targeted shape.11 It has been shown that process temperatures higher than 600 °C can result in the shrinkage and closing of mesopores.12 The secondary reunion of WC nanoparticles is unavoidable during the high temperature carburization process (800–1000 °C),13 which is the main obstacle in the preparation of nanosized WC materials.
Herein, we report for the first time, the solvent- and template-free synthesis of nanosized core–shell WC@meso-Pt materials via a gas–solid reaction (Fig. S1, ESI†), followed by a simple Pt replacement of Cu.
The microsized ammonium metatungstate hydrate precursor ((NH4)2W4O13·xH2O, approximately 12 μm in size) was mixed with copper nitrate by spray drying and then self-disintegrated into nanosized particles in a temperature programmed gas–solid reaction (Fig. S2, ESI†). The phase purity of the obtained samples was characterized using a Thermo ARL SCINTAG X'TRA diffractometer with Cu Kα radiation (λ of 0.1541 nm). As shown in Fig. 1a, the intensive diffraction patterns of the WC@Cu sample (as-prepared) can be indexed to that of the hexagonal phase (HCP, space group P6m2) with lattice constants a and c of 2.91 Å and 2.84 Å, respectively, for WC (peak intensities and positions are shown and confirmed from JCPDS card number 25-1047). In addition, the diffraction peaks are assigned to the face-centred cubic (fcc, space group Fm3m) Cu crystal, which matches well with the JCPDS card number 04-0836.
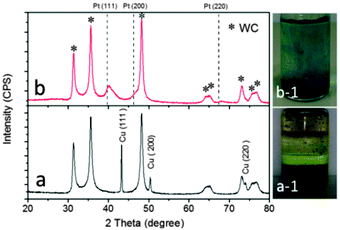 | ||
| Fig. 1 The powder X-ray diffraction (XRD) patterns of (a) the WC@Cu sample and (b) the WC@(Cu)Pt sample obtained from WC@Cu after the reaction of Pt replacing Cu in a chloroplatinic acid solution. | ||
The WC@Cu powders were then treated by a reaction of Pt replacing Cu in a chloroplatinic acid (H2PtCl6) solution (Fig. 1a–1) at 50 °C. It can be seen clearly that the replacement reaction changes the colour of the solution from yellow (Pt4+ ions) to blue (Cu2+ ions) (Fig. 1b–1). The resultant solid powder sample was separated and characterized by XRD (Fig. 1b). The diffraction peaks at 2θ, approximately 39.7°, 46.2° and 67.4° in curve b (Fig. 1b), were assigned to (111), (200) and (220) facets of the fcc Pt crystal, respectively. Careful examination of the Pt(111) peak showed that there was a shift to a slightly higher 2θ value of approximately 40.3°, which may indicate incorporation of trace smaller Cu atoms into the Pt lattice resulting in lattice contraction.14 Energy dispersive X-ray spectroscopy (EDS) data (see Fig. S3, ESI†) confirm the presence of trace Cu in the as-prepared sample, named WC@(Cu)Pt.
It is worth noting that copper nitrate was introduced in the precursor mixture to prevent reunion of the WC nanoparticles. Size-controlled growth is crucial to producing well-defined nanoparticles; self-assembly is a cost-effective way to achieve this purpose. The reunion-restriction of the WC nanoparticles relies on the addition of copper nitrate; Cu(NO3)2 decomposition could produce gas which could separate the WC particles.5bFig. 2 shows the scanning electron microscopy (SEM) images of the resultant nanosized WC@(Cu)Pt, in comparison to the microsized precursor. In a parallel experiment, in which the copper nitrite was omitted from a precursor mixture, only microsized irregular particles were obtained (Fig. S4, ESI†).
 | ||
| Fig. 2 Scanning electron microscopy (SEM) images of (a) the spray dried precursor and (b) the nanosized WC@(Cu)Pt sample, the inset shows particle size distribution. | ||
During the heating process, ammonium metatungstate decomposed to WO3 (Fig. S5, ESI†) which has a much higher melting point of 1473 °C. The phase separation could result in a core–shell structure (Fig. S6 and S7, ESI†). At reaction temperatures of 350–800 °C, reduction and carburization of CuO and WO3 would take place synchronously and the WC@Cu core–shell nanoballs (Fig. S7, ESI†) were formed. Formation of WC may follow the route (eqn (1)) given below.15
| Precursor → WO3 → WO2 → W2C → WC | (1) |
At 380 °C, the pressure in the furnace tube fluctuated due to the significant consumption of H2 and the complete reduction of CuO to Cu (eqn (2)).
| CuO + H2/CO → Cu + H2O/CO2 | (2) |
Pt replacement of Cu can take place spontaneously because the standard reduction potential of the PtCl62−/Pt pair (1.188 V vs. standard hydrogen electrode (SHE)) is much higher than that of the Cu2+/Cu pair (0.34 V vs. SHE), resulting in Pt4+ being reduced to Pt, whilst Cu being oxidized to Cu2+.15 The EDS data obtained from the WC@(Cu)Pt reveal that there is 9.62% Pt, 0.26% Cu and 90.11% WC by weight percentage (Fig. S3, ESI†). Transmission electron microscopy (TEM) (Fig. 3a and b) images of WC@(Cu)Pt display nanoballs with core–shell structure. The core–shell structure was further confirmed by EDS line-scan analysis (Fig. 3c) of the profiles of Cu, Pt and W along the selected line across the particle.
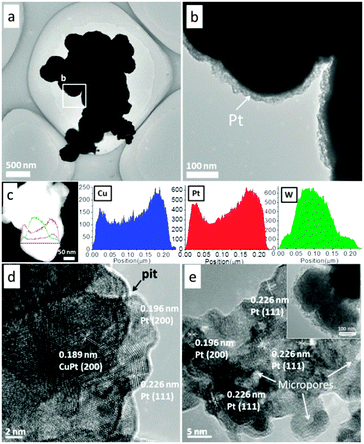 | ||
| Fig. 3 Transmission electron microscopy (TEM) images (a and b), EDS line-scan analysis (c) and high resolution transmission electron microscopy (HRTEM) image (d) of the as-prepared WC@(Cu)Pt sample; and HRTEM image (e) of the WC@meso-Pt sample obtained after performing cyclic voltammetry for WC@(Cu)Pt in an acidic methanol solution. | ||
The lattice spacings for the Pt shell area were found to be 2.26 Å and 1.96 Å (Fig. 3d), which match well with the lattice spacing of (111) and (200) planes of the Pt crystal. For the inner area of the shell, the measured lattice spacing of 1.89 Å is very close to the value obtained for the cubic lattice of CuPt. Small pits appearing on the outer surface of the particle suggest that the corrosion process must have been locally initiated rather than all over the surface. Dissolved chloride (Cl−) ions (coming from H2PtCl6) may play a significant role in this corrosion process.16 The adsorption of Cl− ions on the surface is likely to enhance the rate of surface diffusion and thus can speed up the pitting process which may also favour the formation of mesoporous Pt.
For application in direct methanol fuel cells (DMFCs), the electrocatalytic activity of WC@(Cu)Pt towards methanol oxidation was investigated. The cyclic voltammograms (CVs) were collected in 2 M CH3OH + 1 M H2SO4solution. Interestingly, it was found that the first and second cycles were unstable and some peaks disappeared with additional cycling (Fig. S8a, ESI†). The peak centred at around 0.20 V in the initial two forward scans was the result of the dissolution/oxidation of Cu which was verified by the CVs obtained from the WC@Cu sample (Fig. S8b, ESI†). The morphology of the electrooxidation treated WC@(Cu)Pt sample was then checked, and no significant change in the particle size (see inset in Fig. 3e) was found. However, the dispersion of the Pt shell appears to have a significantly higher permeability.
For the as-prepared WC@(Cu)Pt, Pt(111), Pt(200) and a trace amount of CuPt(200) phases are dispersed in the shell (Fig. 3d), whereas for the electrooxidation treated sample, no Cu was detected and almost all Pt particles were found to have micropores (Fig. 3e). This is because, as observed in the forward scans in the CVs (Fig. S8a, ESI†), the oxidative removal of Cu caused the pits to form on the surface of the particles, resulting in the formation of micropores on the Pt layer. The treated sample which exhibits mesoporous Pt shell structure was identified as WC@m-Pt, with an effective Pt crystal plane (111) in the shell area, displaying an expanded surface area and a meso/microporous structure (Fig. 3e, pores (∼2 nm), also see the pore-size distribution and small angle-XRD data in Fig. S9, ESI†).
Further electrochemical tests show that, compared with the conventional Pt/C(10 wt% Pt, Johnson Matthey Corp. through Hesen Co.), the electrochemically active surface area (evidenced from the hydrogen adsorption–desorption current density in an acid electrolyte, see the inset in Fig. 4a) of the WC@m-Pt is much higher, and its electrocatalytic activity towards methanol electrooxidation is significantly higher than that of Pt/C, e.g., a peak current density of 275 mA cm−2 in comparison with 136 mA cm−2 observed in the CVs (Fig. 4a).
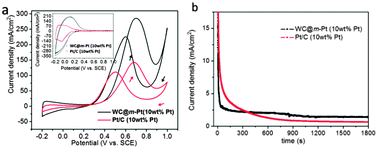 | ||
| Fig. 4 (a) CVs of the WC@m-Pt and Pt/C in 2 M CH3OH + 1 M H2SO4 solution at a scan rate of 50 mV s−1 at 25 °C; the inset in (a) shows CVs in 1 M H2SO4 supporting electrolyte. (b) Chronoamperometric data (current-time profiles) of these samples at 0.3 V in 2 M CH3OH + 1 M H2SO4 solution. | ||
In addition to activity, stability of the electrocatalyst is also an important feature for its application. Compared to Pt/C, WC@m-Pt exhibited a superior long-term electrocatalytic activity towards methanol electrooxidation, as evidenced from the chronoamperometric data (Fig. 4b) which show a stable and significantly higher current density at 0.3 V after 30 min, revealing the inherent catalytic stability. The mesostructure of the Pt-shell (crystallite size around 2.8 nm, calculated by the Scherrer formula) may facilitate the interaction between the WC core and Pt shell and the synergy effect on methanol oxidation. The detailed mechanistic studies are underway and will be reported in a separate paper.
In summary, we have developed a facile method to synthesize core–shell WC@meso-Pt nanocatalysts with a porous shell morphology by carburizing the micro-sized ammonium metatungstate precursor mixed with copper nitrate via a typical gas–solid reaction, followed by simple Pt replacement of the Cu and electrochemical treatment. The mesoporous nanocomposite displays higher catalytic activity and long-term stability towards the methanol electrooxidation reaction than the commercial Pt/C catalyst. The current methods can be extended or adapted to synthesize other noble metals and their alloy nanocomposites on transition metal carbides for wider ranges of electrocatalysis or heterogeneous catalysis.
This work was financially supported by International Science & Technology Cooperation Program of China (2010DFB63680), Queen's University Belfast and EPSRC (EP/I013229/1). C.M. and X.L. thank QUB for the awards of the titles of Visiting Professor and Visiting Research Associate, respectively.
Notes and references
- (a) H.-C. Youn, S. Baral and J. H. Fendler, J. Phys. Chem., 1988, 92, 6320 CrossRef CAS; (b) A. Henglein, Chem. Rev., 1989, 89, 1861 CrossRef CAS.
- (a) C. F. Hoener, K. A. Allan, A. J. Bard, A. Campion, M. A. Fox, T. E. Malluok, S. E. Webber and J. M. White, J. Phys. Chem., 1992, 96, 3812 CrossRef CAS; (b) O. O. Fashedemi, B. Juliesb and K. I. Ozoemena, Chem. Commun., 2013, 49, 2034 RSC.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373 CrossRef PubMed.
- (a) F. F. P. Medeiros, S. A. De Oliveira, C. P. De Souza, A. G. P. Da Silva, U. U. Gomes and J. F. De Souza, Mater. Sci. Eng., A, 2001, 315, 58 CrossRef; (b) R. J. Wrobel, W. Arabczyk and U. Narkiewicz, Cryst. Res. Technol., 2012, 47, 1164 CrossRef CAS.
- (a) C. W. Wu, K. Sullivan, S. Chowdhury, G. Q. Jian, L. Zhou and M. R. Zachariah, Adv. Funct. Mater., 2012, 22, 78 CrossRef CAS; (b) G. Q. Jian, L. Liu and M. R. Zachariah, Adv. Funct. Mater., 2013, 23, 1341 CrossRef CAS; (c) Z. C. Sun, Q. Zhou and L. S. Fan, Langmuir, 2013, 29, 12520 CrossRef CAS PubMed; (d) A. Prakash, A. V. McCormick and M. R. Zachariah, Nano Lett., 2005, 5, 1357 CrossRef CAS.
- J. P. Bosco, K. Sasaki, M. Sadakane, W. Ueda and J. G. G. Chen, Chem. Mater., 2010, 22, 966 CrossRef CAS.
- (a) H. Böhm, Nature, 1970, 227, 483 CrossRef PubMed; (b) R. B. Levy and M. Boudart, Science, 1973, 181, 547 CAS; (c) E. C. Weigert, A. L. Stottlemyer, M. B. Zellner and J. G. Chen, J. Phys. Chem. C, 2007, 111, 14617 CrossRef CAS.
- (a) M. Boudart and L. D. Ptak, J. Catal., 1970, 16, 90 CrossRef CAS; (b) C. A. Ma, Z. Y. Chen, W. F. Lin, F. M. Zhao and M. Q. Shi, Microporous Mesoporous Mater., 2012, 149, 76 CrossRef CAS PubMed.
- (a) R. H. Wang, Y. Xie, K. Y. Shi, J. Q. Wang, C. G. Tian, P. K. Shen and H. G. Fu, Chem.–Eur. J., 2012, 18, 7443 CrossRef CAS PubMed; (b) M. K. Jeon, K. R. Lee, W. S. Lee, H. Daimon, A. Nakahara and S. I. Woo, J. Power Sources, 2008, 185, 927 CrossRef CAS PubMed.
- Z. Q. Niu, D. S. Wang, R. Yu, Q. Peng and Y. D. Li, Chem. Sci., 2012, 3, 1925 RSC.
- (a) CN pat., 1 0153606.8, 2009 Search PubMed; (b) CN pat., 1 0154483.X, 2009 Search PubMed.
- Z. Y. Chen, M. Q. Shi, C. A. Ma, Y. Q. Chu and A. J. Zhu, Powder Technol., 2013, 235, 467 CrossRef CAS PubMed.
- (a) C. S. Pang, Z. M. Guo, J. Luo, T. Hou and J. Bing, Int. J. Refract. Met. Hard Mater., 2010, 28, 394 CrossRef CAS PubMed; (b) A. Kumar, K. Singh and O. P. Pandey, Physica E, 2010, 42, 2477 CrossRef CAS PubMed.
- D. Liang, J. Gao, J. H. Wang, P. Chen, Y. F. Wei and Z. Y. Hou, Catal. Commun., 2011, 12, 1059 CrossRef CAS PubMed.
- (a) M. Mohl, D. Dobo, A. Kukovecz, Z. Konya, K. Kordas, J. Q. Wei, R. Vajtai and P. M. Ajayan, J. Phys. Chem. C, 2011, 115, 9403 CrossRef CAS; (b) S. Zhou, B. Varughese, B. Eichhorn, G. Jackson and K. Mcllwrath, Angew. Chem., Int. Ed., 2005, 44, 4539 CrossRef CAS PubMed.
- R. C. Newman and K. Sieradzki, Science, 1994, 263, 1708 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD patterns and SEM images of the spray dried precursor, TEM and EDS line-scan analysis of WC@Cu, EDS of WC@(Cu)Pt, CVs of WC@(Cu)Pt and WC@Cu in methanol and acid solutions, nitrogen adsorption isotherm and pore size distribution, BET and XRD analysis of as-prepared WC@m-Pt. See DOI: 10.1039/c3cc46595k |
| This journal is © The Royal Society of Chemistry 2013 |