 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogenase biomimetics: Fe2(CO)4(μ-dppf)(μ-pdt) (dppf = 1,1′-bis(diphenylphosphino)ferrocene) both a proton-reduction and hydrogen oxidation catalyst†
Shishir
Ghosh
a,
Graeme
Hogarth
*ab,
Nathan
Hollingsworth
a,
Katherine B.
Holt
*a,
Shariff E.
Kabir
c and
Ben E.
Sanchez
a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: g.hogarth@ucl.ac.uk
bDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London SE1 1DB, UK. E-mail: Graeme.hogarth@kcl.ac.uk
cDepartment of Chemistry, Jahangirnagar University, Savar, Dhaka 1342, Bangladesh
First published on 29th November 2013
Abstract
Fe2(CO)4(μ-dppf)(μ-pdt) catalyses the conversion of protons and electrons into hydrogen and also the reverse reaction thus mimicing both types of binuclear hydrogenase enzymes.
Hydrogenases are enzymes capable of reversibly converting protons and electrons into hydrogen1 and over the past two decades their active sites have been discerned primarily from crystallographic studies,2–4 with three phylogenetically different enzyme types being identified. The two most widely studied of these are the so-called [FeFe]H2ase and [NiFe]H2ase enzymes (Chart 1) the active sites of which contain two transition metal atoms. While both enzyme types are able to catalyse both the reduction of protons and oxidation of hydrogen, [FeFe]H2ase enzymes are more efficient with respect to the former, while [NiFe]H2ase enzymes are favoured for the oxidation of hydrogen.5
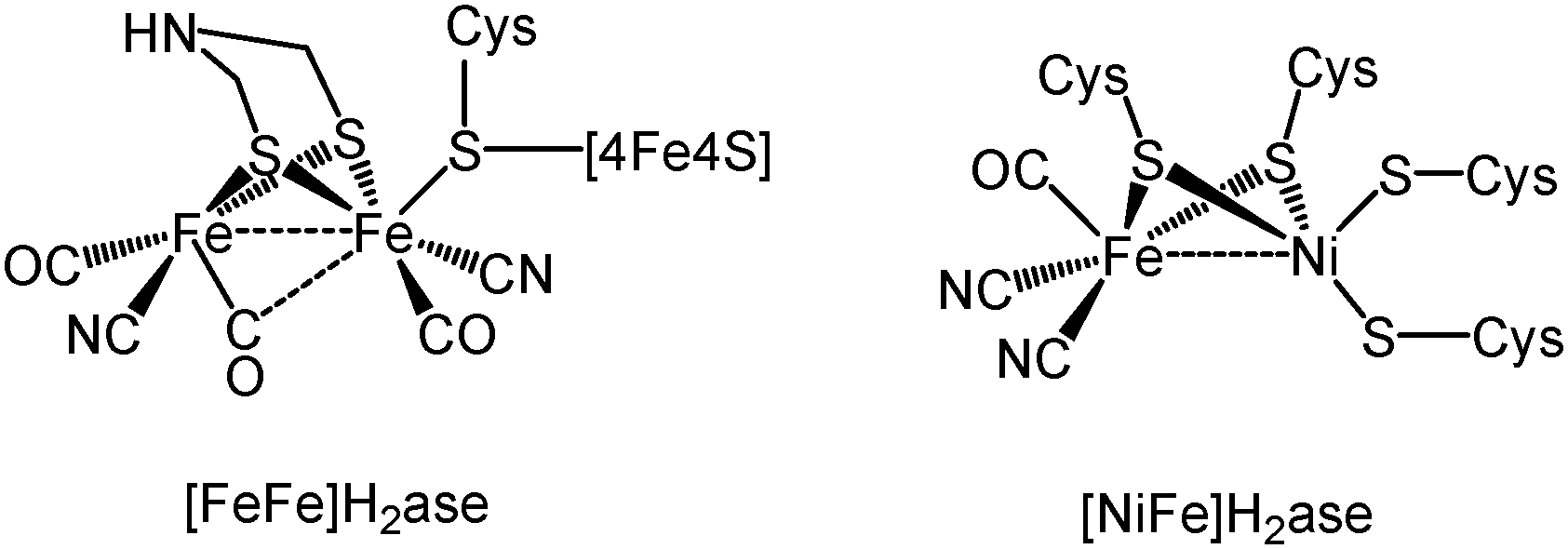 | ||
| Chart 1 Structure of the active site of [FeFe]H2ase and [NiFe]H2ase. | ||
Over the past 15 years, a range of structural and functional biomimetics of the active sites of these enzymes have studied5,6 with key insights into the likely mechanism(s) being determined7 and recently some impressive turnovers for the electrocatalytic reduction of protons being reported.8 However, as far as we are aware, no biomimetic has yet been shown to both be catalytic for the reduction of protons and electrons to hydrogen and also the oxidation of hydrogen to protons and electrons.9 We herein describe Fe2(CO)4(μ-dppf)(μ-pdt) (2) {pdt = S(CH2)3S, dppf = 1,1′-bis(diphenylphosphino)ferrocene} which we have shown is a catalyst for both of these transformations.
Heating a toluene solution of equimolar amounts of Fe2(CO)6(μ-pdt) (1) with dppf initially leads to formation of the linked tetranuclear complex {Fe2(CO)5(μ-pdt)}2(μ,κ1,κ1-dppf)10 and unreacted dppf, which slowly rearranges to afford Fe2(CO)4(μ-dppf)(μ-pdt) (2) in moderate yields‡ as an air-stable orange solid (Scheme 1) being characterised by analytical and spectroscopic data together with a single-crystal X-ray diffraction study, the results of which are summarised in Fig. 1. The molecule is as expected and bond lengths and angles are generally within the ranges of those seen in related complexes.10,11 The iron–iron bond length of 2.6133(6) Å is, however, some 0.1 Å longer than is generally the case10–12 suggesting that the flexible nature of the dppf ligand allows this bond to relax. The non-bonding iron–iron distances of 4.581 and 4.613 Å suggest that there is no direct contact between the two redox centres in the molecule.
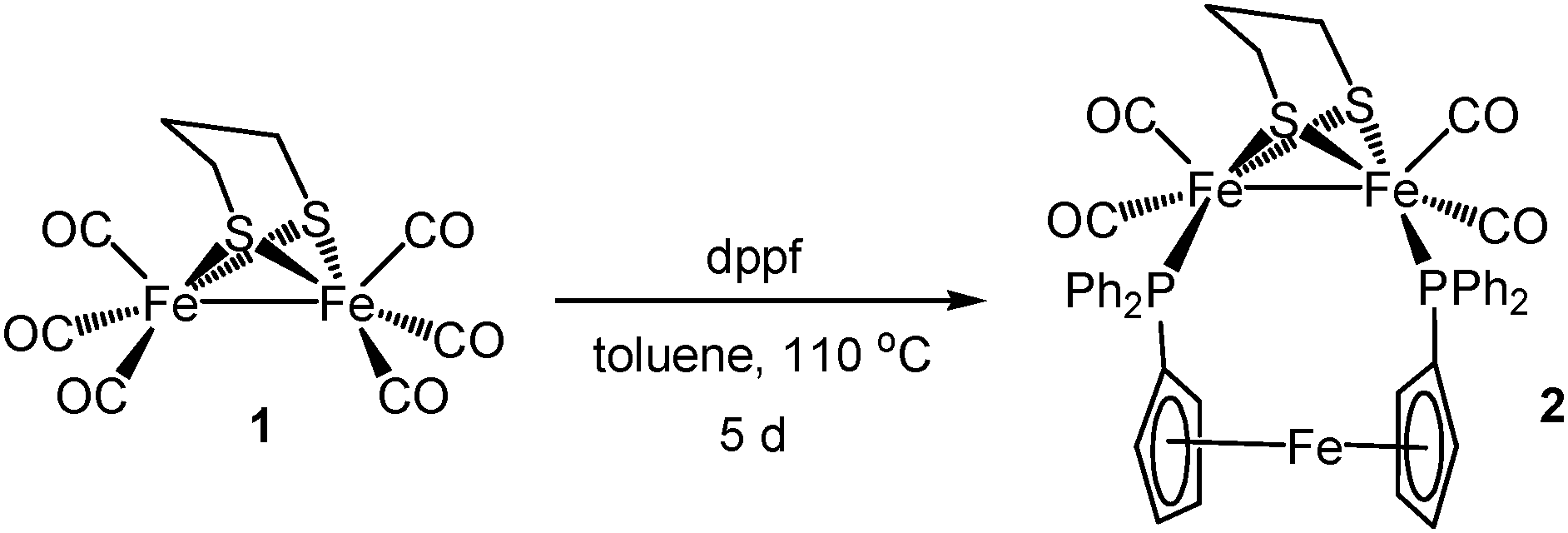 | ||
| Scheme 1 Preparation of Fe2(CO)4(μ-dppf)(μ-pdt) (2). | ||
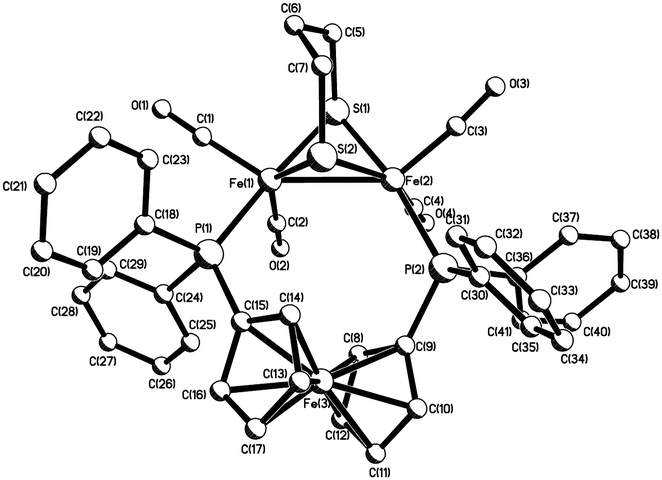 | ||
| Fig. 1 Molecular structure of 2. Selected bond lengths [Å] and bond angles [°]: Fe(1)–Fe(2) 2.6133(6), Fe(1)–P(1) 2.2256(6), Fe(2)–P(2) 2.2679(6), P(1)–Fe(1)–S(1) 174.34(2), P(2)–Fe(2)–S(1) 167.79(2). | ||
Cyclic voltammograms (CVs) of 2 were recorded in acetonitrile at various scan rates as shown in Fig. 2. The complex undergoes an electrochemically reversible oxidation at E1/2 = 0.05 V (ΔE = 60 mV) and a further reversible oxidation at E1/2 = 0.685 V (ΔE = 70 mV). The former is associated with oxidation of the diiron centre and the latter most likely with the ferrocene moiety (see later). The reversibility of both oxidative processes is maintained at all scan rates. The complex also shows two overlapping irreversible reduction peaks at Ep = −2.10 V and Ep = −2.19 V which become separated at higher scan rates (≥0.25 V s−1) (Fig. 2). Two small oxidation peaks are also observed at Ep = −1.80 V and Ep = −1.53 V on the return scan being due to the product formed in the reductive processes, whilst the small reduction peak appeared at Ep = −0.35 V on the return scan is associated with the first oxidation product.
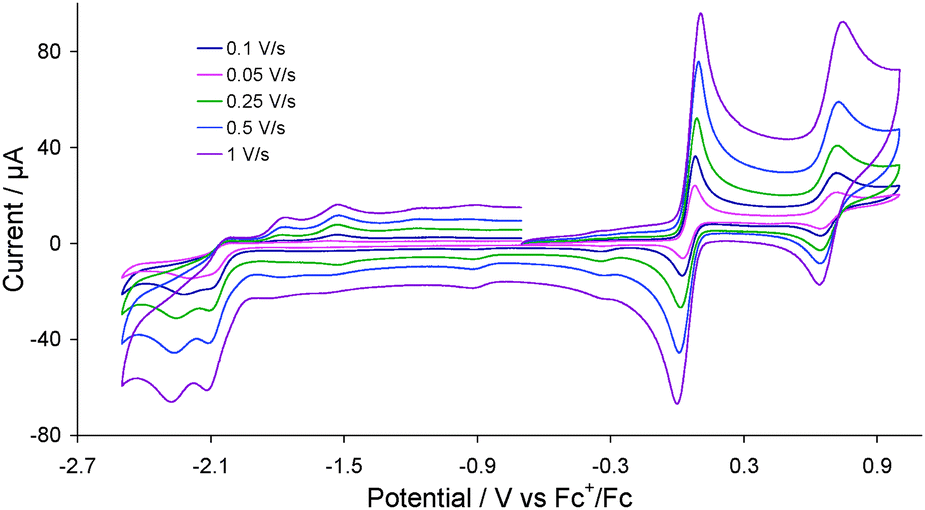 | ||
| Fig. 2 CVs of 2 in MeCN (1 mM solution, supporting electrolyte [NBu4][PF6], glassy carbon electrode, potential vs. Fc+/Fc) at various scan rates. | ||
Upon chemical oxidation, by addition of FcPF6 to a CH2Cl2 solution of 2, new IR absorption bands appear at 2044 and 2013 cm−1 (Fig. S1, ESI†). The 60 cm−1 shift of the first νCO band to higher energy is indicative of oxidation of the diiron centre allowing assignment of the couple at 0.05 V vs. Fc+/Fc to this process. The second oxidation we believe is associated with the dppf ligand. Uncoordinated dppf undergoes an irreversible oxidation at 0.20 V which becomes reversible and shifts to more positive potentials upon coordination to a metal centre.13 The relative position of the second oxidative process vs. Fc+/Fc and its chemical reversibility is consistent with the FeII/III couple of the ferrocene moiety.
We next assessed the ability of 2 to bind a proton. Addition of one molar equivalent of HBF4·Et2O to a CH2Cl2 solution of 2 (or two to an MeCN solution) resulted in the rapid and clean formation of the cationic-hydride [Fe2(CO)4(μ-H)(μ-dppf)(μ-pdt)][BF4] (3).‡ Further, and unlike most related cationic complexes,11 addition of base leads to regeneration of the neutral complex. This suggests that while 2 is able to bind a proton, it is relatively weakly held. Related Fe2(CO)4(μ-diphosphine)(μ-dithiolate) complexes do not generally form stable cationic hydrides, the exceptions being Fe2(CO)4(μ-Cy2PCH2PCy2)(μ-pdt) and Fe2(CO)4{μ-Ph2P(CH2)4PPh2}(μ-pdt) containing basic and flexible diphosphines respectively.11 Thus, the greater flexibility of dppf in 2 appears to be the reason for its ability to bind a proton.
Complex 2 was first tested as a proton reduction catalyst in the presence of HBF4·Et2O in MeCN. Fig. 3 shows the CVs upon addition of between 1–10 equivalents of acid. A new reduction wave appears at Ep = −1.70 V upon addition of acid being associated with reduction of 3, its height growing with increasing amounts of acid, being characteristic of electrocatalytic proton reduction.6 At higher amounts of acid (≥7 molar equivalents) this wave splits into two distinct peaks possibly resulting from reduction of the putative cation [HFe2(CO)4(μ-H)(μ-dppf)(μ-pdt)]+ (see below). Another catalytic wave is also observed at Ep = −2.10 V which competes with the direct reduction of HBF4·Et2O by the glassy carbon electrode as this electrode becomes catalytically active beyond −2.00 V in presence of strong acids.14 On the return scan a further catalytic wave is seen at Ep = −1.55 V implying that the species responsible for the first catalytic wave is regenerated. Thus it appears that 2 enters into the catalytic cycle via a CE mechanism to generate the neutral paramagnetic complex Fe2(CO)4(μ-H)(μ-dppf)(μ-pdt)15 which either protonates or undergoes a further reduction before second protonation to liberate hydrogen. The peak heights of the oxidative processes do not change during the experiment showing the robustness of 2 under the operating conditions.
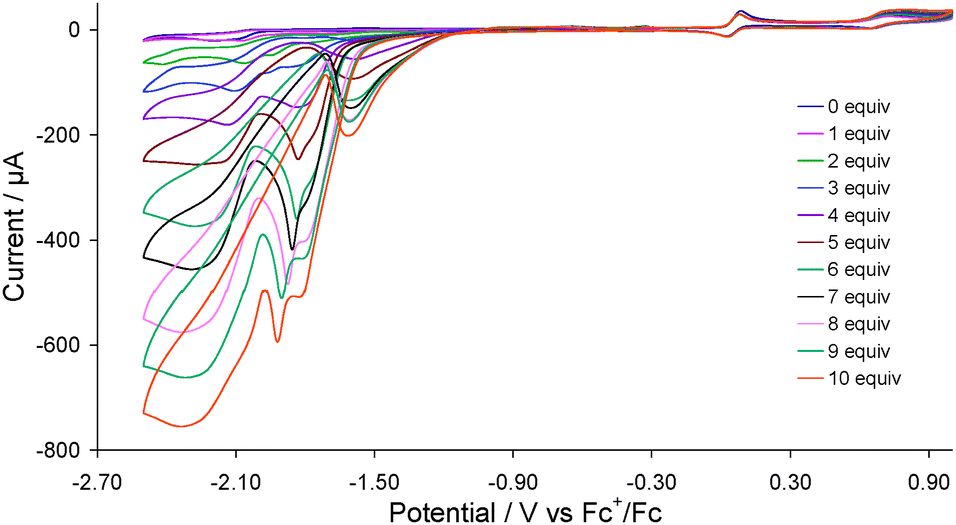 | ||
| Fig. 3 CVs of 2 in the absence and presence of 1–10 molar equivalents of HBF4·Et2O (1 mM solution in MeCN, supporting electrolyte [NBu4][PF6], scan rate 0.1 V s−1, glassy carbon electrode, potential vs. Fc+/Fc). | ||
Recent developments in hydrogenase biomimics suggest that H2 activation can be favoured by the presence of a mild and chemically inert oxidant in the diiron models.16 The concept was recently experimentally implemented by Camara and Rauchfuss17,18 who utilised (C5Me5)Fe(C5Me4)CH2PEt2 (FcP*) as the intramolecular oxidant, the FeII/III couple (E1/2 = −0.59 V) of which lies closer to the H2/H+ couple vs. the Fc+/Fc couple.18 They showed that the dication of Fe2(CO)3(κ2-Ph2PCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPPh2)(κ1-FcP*){μ-SCH2N(Bz)CH2S} (A) cleaves H2, being facilitated by an intramolecular electron-transfer in its doubly oxidised state, the electron transferring from the diiron unit to the pendent FcP* ligand i.e. switching from Fe(III)Fe(II)Fe(I) to Fe(II)Fe(II)Fe(II).18 In contrast, an analogue of A in which FcP* is replaced by PMe3 is catalytically inactive towards H2 oxidation.18 That there is electronic communication between the diiron core and the ferrocene in A despite the presence of a methylene linker unit prompted us to investigate the possibility of electronic communication between the two redox-active metal centres in 2. Indeed we found that 2 catalytically cleaves H2 in presence of a base (pyridine) in its 22+ state (Fig. 4). Thus, addition of equimolar amount of pyridine to an acetonitrile solution of 2 under H2 results in an increase of the oxidative peak current of the second oxidation process of 2 by 10 μA, which reaches 22 μA upon addition of 10 equivalents of pyridine. No such catalytic wave was observed when the same experiment was carried out in absence of base (Fig. S5, ESI†) or H2 (Fig. S6, ESI†). Similarly Fe2(CO)4(μ-Ph2PCH2PPh2)(μ-pdt)11 does not show catalytic waves under the same conditions even when ferrocene is added. At this stage we do not have a clear view of the likely mechanism operating. It has been proposed18 and examined theoretically19 that A2+ heterolytically cleaves H2 to afford a terminal hydride and nitrogen-bound proton. This clearly cannot occur in the case of 2 and thus we tentatively propose the intermediate formation of a cationic dihydride.
CHPPh2)(κ1-FcP*){μ-SCH2N(Bz)CH2S} (A) cleaves H2, being facilitated by an intramolecular electron-transfer in its doubly oxidised state, the electron transferring from the diiron unit to the pendent FcP* ligand i.e. switching from Fe(III)Fe(II)Fe(I) to Fe(II)Fe(II)Fe(II).18 In contrast, an analogue of A in which FcP* is replaced by PMe3 is catalytically inactive towards H2 oxidation.18 That there is electronic communication between the diiron core and the ferrocene in A despite the presence of a methylene linker unit prompted us to investigate the possibility of electronic communication between the two redox-active metal centres in 2. Indeed we found that 2 catalytically cleaves H2 in presence of a base (pyridine) in its 22+ state (Fig. 4). Thus, addition of equimolar amount of pyridine to an acetonitrile solution of 2 under H2 results in an increase of the oxidative peak current of the second oxidation process of 2 by 10 μA, which reaches 22 μA upon addition of 10 equivalents of pyridine. No such catalytic wave was observed when the same experiment was carried out in absence of base (Fig. S5, ESI†) or H2 (Fig. S6, ESI†). Similarly Fe2(CO)4(μ-Ph2PCH2PPh2)(μ-pdt)11 does not show catalytic waves under the same conditions even when ferrocene is added. At this stage we do not have a clear view of the likely mechanism operating. It has been proposed18 and examined theoretically19 that A2+ heterolytically cleaves H2 to afford a terminal hydride and nitrogen-bound proton. This clearly cannot occur in the case of 2 and thus we tentatively propose the intermediate formation of a cationic dihydride.
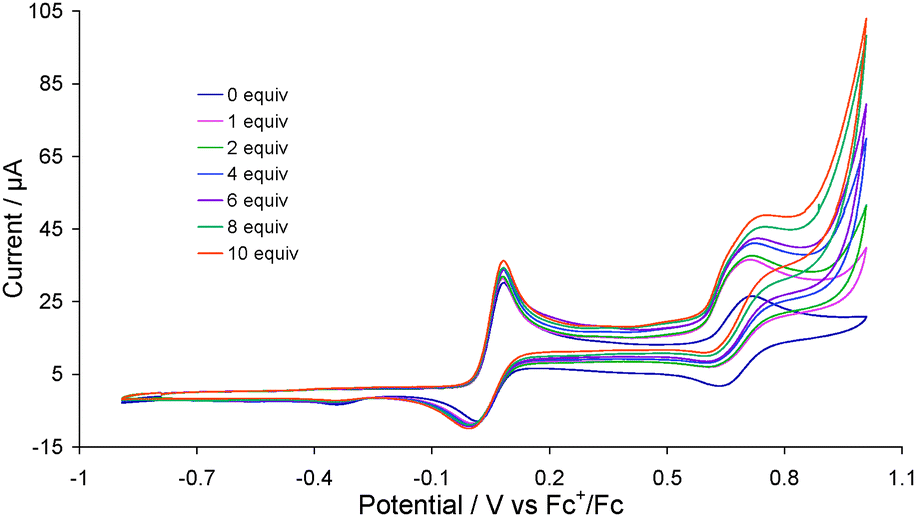 | ||
| Fig. 4 CVs of 2 in the absence of pyridine and in the presence of varying molar equivalents of pyridine under a H2 atmosphere (1 mM solution in MeCN, supporting electrolyte [NBu4][PF6], scan rate 0.1 V s−1, glassy carbon electrode, potential vs. Fc+/Fc). | ||
In summary we have shown that a biomimetic of the diiron hydrogenase can catalyse both the reduction of protons and H2 oxidation. We are currently developing a range of related biomimetics containing different secondary redox-active centres20 and using density functional theory calculations in order to more fully understand the electronic structure of 22+ and the nature of the H2 oxidation process.
We are grateful to the Commonwealth Scholarship Commission for the award of a Commonwealth Scholarship to S.G. and the EPSRC for a postdoctoral fellowship to N.H.
Notes and references
- M. W. W. Adams and E. I. Stiefel, Science, 1998, 282, 1842–1843 CrossRef CAS; R. Cammack, Nature, 1999, 397, 214–215 CrossRef PubMed; M. Frey, ChemBioChem, 2002, 3, 153–160 CrossRef.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS; Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecillacamps, Structure, 1999, 7, 13–23 CrossRef.
- A. Volbeda, M. H. Charon, C. Piras, C. E. Hatchikian, M. Frey and J. C. Fontecillacamps, Nature, 1995, 373, 580–587 CrossRef CAS PubMed.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS PubMed; S. Shima, E. J. Lyon, R. K. Thauer, B. Mienert and E. Bill, J. Am. Chem. Soc., 2005, 127, 10430–10435 CrossRef PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- For some reviews of this area see: I. P. Georgakaki, L. M. Thomson, E. J. Lyon, M. B. Hall and M. Y. Darensbourg, Coord. Chem. Rev., 2003, 238–239, 255–266 CrossRef CAS; D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268–287 RSC; T. B. Rauchfuss, Inorg. Chem., 2004, 43, 14–26 CrossRef PubMed; L. Sun, B. Åkermark and S. Ott, Coord. Chem. Rev., 2005, 249, 1653–1663 CrossRef PubMed; X. Liu, S. K. Ibrahim, C. Tard and C. J. Pickett, Coord. Chem. Rev., 2005, 249, 1641–1652 CrossRef PubMed; J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2005, 249, 1664–1676 CrossRef PubMed.
- C. Greco, M. Bruschi, L. D. Gioia and U. Ryde, Inorg. Chem., 2007, 46, 5911–5921 CrossRef CAS PubMed; C. Greco, M. Bruschi, P. Fantucci, U. Ryde and L. De Gioia, J. Am. Chem. Soc., 2011, 133, 18742–18749 CrossRef PubMed; S. Trohalaki and R. Pachter, Int. J. Hydrogen Energy, 2010, 35, 5318–5331 CrossRef PubMed.
- S. Dey, A. Rana, S. G. Day and A. Dey, ACS Catal., 2013, 3, 429–436 CrossRef CAS.
- N. Wang, M. Wang, L. Chen and L. Sun, Dalton Trans., 2013, 42, 12059–12071 RSC.
- X.-F. Liu and B.-S. Yin, J. Coord. Chem., 2010, 63, 4061–4067 CrossRef CAS.
- F. I. Adam, G. Hogarth, S. E. Kabir and I. Richards, C. R. Chim., 2008, 11, 890–905 CrossRef CAS PubMed; F. I. Adam, G. Hogarth and I. Richards, J. Organomet. Chem., 2007, 692, 3957–3968 CrossRef PubMed.
- S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt, I. Richard, M. G. Richmond, B. Sanchez and D. Unwin, Dalton Trans., 2013, 42, 6775–6792 RSC; F. Ridley, S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt and D. Unwin, J. Electroanal. Chem., 2013, 703, 14–22 CrossRef CAS PubMed; G. Hogarth, S. E. Kabir and I. Richards, Organometallics, 2010, 29, 6559–6568 CrossRef; L.-C. Song, C.-G. Li, J.-H. Ge, Z.-Y. Yang, H.-T. Wang, J. Zhang and Q.-M. Hu, J. Inorg. Biochem., 2008, 102, 1973–1979 CrossRef PubMed; W. Gao, J. Ekström, J. Liu, C. Chen, L. Eriksson, L. Weng, B. Åkermark and L. Sun, Inorg. Chem., 2007, 46, 1981–1991 CrossRef PubMed.
- D. L. DuBois, C. W. Eigenbrot, J. A. Miedaner, J. C. Smart and R. C. Haltiwanger, Organometallics, 1986, 5, 1405–1411 CrossRef CAS; B. D. Swartz and C. Nataro, Organometallics, 2005, 24, 2447–2451 CrossRef; C. Nataro, A. N. Campbell, M. A. Ferguson, C. D. Incarvito and A. L. Rheingold, J. Organomet. Chem., 2003, 673, 47–55 CrossRef; G. Pilloni, B. Longato and B. Corain, J. Organomet. Chem., 1991, 420, 57–65 CrossRef; B. Corain, B. Longato and G. Favero, Inorg. Chim. Acta, 1989, 157, 259–266 CrossRef.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- A. Jablonskyté, J. A. Wright, S. A. Fairhurst, J. N. T. Peck, S. K. Ibrahim, V. S. Oganesyan and C. J. Pickett, J. Am. Chem. Soc., 2011, 133, 18606–18609 CrossRef PubMed.
- J. C. Gordon and G. J. Kubas, Organometallics, 2010, 29, 4682–4701 CrossRef CAS; C. Greco, G. Zampella, L. Bertini, M. Bruschi, P. Fantucci and L. De Gioia, Inorg. Chem., 2007, 46, 108–116 CrossRef PubMed; C. Greco and L. D. Gioia, Inorg. Chem., 2011, 50, 6987–6995 CrossRef PubMed.
- J. M. Camara and T. B. Rauchfuss, J. Am. Chem. Soc., 2011, 133, 8098–8101 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed.
- C. Greco, Inorg. Chem., 2013, 52, 1901–1908 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data. CCDC 956914. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc46456c |
‡ Synthesis of 2. A mixture of 1 (0.10 g, 0.26 mmol) and dppf (0.14 g, 0.26 mmol) in toluene (100 ml) was heated at reflux for 5 d resulting in a colour change from orange to red-brown. After cooling to room temperature, volatiles were removed under reduced pressure to give a dark oily red residue. This was washed with hexanes (3 × 5 ml) and dried. Extraction into a minimum volume of dichloromethane followed by addition of hexanes and rotary evaporation gave 2 as a dry red solid (0.12 g, 52%). 2 can also be prepared upon heating a mixture of {Fe2(CO)5(μ-pdt)}2(μ,κ1,κ1-dppf)10 and dppf in toluene over a similar period. IR ν(CO)(CH2Cl2) 1986s, 1949vs, 1918s 1896w cm−1. 1H NMR (CDCl3) δ 8.01 (t, J 8.2, 2H, Ph), 7.67–6.99 (m, 18H, Ph), 4.93 (brs, 2H, CH), 4.46 (s, 2H, CH), 4.44 (s, 2H, CH), 4.01 (s, 2H, CH), 2.60 (br, 2H, CH2), 2.31 (m, 2H, CH2), 2.13 (br, 2H, CH2). 31P{1H}NMR (CDCl3) 51.3 (s) ppm. Elemental analysis calc. for Fe3S2P2O4C41H35·0.5CH2Cl2 (found): C 54.16 (53.41), H 3.81 (3.75). X-ray data for Fe3S2P2O4C41H35·0.5CH2Cl2: red block, dimensions 0.38 × 0.32 × 0.16 mm, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.7365(19), b = 13.149(3), c = 16.654(3) Å, α = 99.609(3), β = 94.376(3), γ = 111.343(3)°, V = 1936.1(7) Å3, Z = 2, F(000) 944, dcalc. = 1.588 g cm−3, μ = 1.411 mm−1. 16 , a = 9.7365(19), b = 13.149(3), c = 16.654(3) Å, α = 99.609(3), β = 94.376(3), γ = 111.343(3)°, V = 1936.1(7) Å3, Z = 2, F(000) 944, dcalc. = 1.588 g cm−3, μ = 1.411 mm−1. 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 reflections were collected, 8886 unique [R(int) = 0.0333] of which 8134 were observed [I > 2.0σ(I)]. At convergence, R1 = 0.0345, wR2 = 0.0911 [I > 2.0σ(I)] and R1 = 0. 0374, wR2 = 0.0929 (all data), for 511 parameters. CCDC number 956914. Synthesis of 3. To a CH2Cl2 (50 ml) solution of 2 (0.05 g, 0.06 mmol) was added a few drops of HBF4. The mixture was stirred at room temperature for 20 min without any noticeable change. Volatiles were removed under reduced pressure and the resulting deep red oily solid washed with a small portion of Et2O to remove excess acid. The remaining solid was dissolved in a minimum amount of CH2Cl2 which was then layered with hexanes. Slow mixing of the solutions afforded 3 (0.04 g, 73%) as a dry red solid. IR ν(CO)(CH2Cl2) 2058s, 2040s, 2002s cm−1. 1H NMR (CDCl3) δ 8.11–7.33 (m, 20H, Ph), 4.74 (s, 2H, CH), 4.68 (s, 2H, CH), 4.49 (s, 2H, CH), 4.32 (s, 2H, CH), 2.86 (br, 2H, CH2), 2.74 (m, 2H, CH2), 2.48 (br, 2H, CH2), −12.40 (t, J 17.6, 1H, μ-H). 31P{1H} NMR (CD2Cl2) 44.8 (s) ppm. 800 reflections were collected, 8886 unique [R(int) = 0.0333] of which 8134 were observed [I > 2.0σ(I)]. At convergence, R1 = 0.0345, wR2 = 0.0911 [I > 2.0σ(I)] and R1 = 0. 0374, wR2 = 0.0929 (all data), for 511 parameters. CCDC number 956914. Synthesis of 3. To a CH2Cl2 (50 ml) solution of 2 (0.05 g, 0.06 mmol) was added a few drops of HBF4. The mixture was stirred at room temperature for 20 min without any noticeable change. Volatiles were removed under reduced pressure and the resulting deep red oily solid washed with a small portion of Et2O to remove excess acid. The remaining solid was dissolved in a minimum amount of CH2Cl2 which was then layered with hexanes. Slow mixing of the solutions afforded 3 (0.04 g, 73%) as a dry red solid. IR ν(CO)(CH2Cl2) 2058s, 2040s, 2002s cm−1. 1H NMR (CDCl3) δ 8.11–7.33 (m, 20H, Ph), 4.74 (s, 2H, CH), 4.68 (s, 2H, CH), 4.49 (s, 2H, CH), 4.32 (s, 2H, CH), 2.86 (br, 2H, CH2), 2.74 (m, 2H, CH2), 2.48 (br, 2H, CH2), −12.40 (t, J 17.6, 1H, μ-H). 31P{1H} NMR (CD2Cl2) 44.8 (s) ppm. |
| This journal is © The Royal Society of Chemistry 2014 |