 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust visible light photoswitching with ortho-thiol substituted azobenzenes†
Subhas Samanta, Theresa M. McCormick, Simone K. Schmidt, Dwight S. Seferos and G. Andrew Woolley*
Department of Chemistry, University of Toronto, 80 St. George St. Toronto, ON M5S 3H6, Canada. E-mail: awoolley@chem.utoronto.ca; Fax: +1-416-978-8775; Tel: +1-416-978-0675
First published on 18th September 2013
Abstract
Introduction of S-ethyl groups in all four ortho positions of azobenzene prevents reduction of the azo group by intracellular glutathione, while enhancing the absorptivity to ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 in the blue and green regions of the visible spectrum. cis-to-trans isomerization occurs thermally on the minutes timescale. Further, this substitution pattern permits switching with red light, a color that is more penetrating through biological tissues than other parts of the visible spectrum.
000 M−1 cm−1 in the blue and green regions of the visible spectrum. cis-to-trans isomerization occurs thermally on the minutes timescale. Further, this substitution pattern permits switching with red light, a color that is more penetrating through biological tissues than other parts of the visible spectrum.
The majority of applications of azobenzenes as molecular switches use UV light to trigger trans-to-cis isomerization.1 In biological systems UV irradiation is highly scattered and can be toxic.2,3 In nanomaterials applications, UV light can be scattered by small structures and is thereby poorly penetrating.4–6 As a consequence there has been considerable interest in developing photoswitches that operate with visible light.4,7–10
Azobenzenes with para donor substituents and push–pull derivatives can show strongly red-shifted π–π* bands but often at the cost of very rapid thermal relaxation (milliseconds) making production of large fractions of the cis isomer difficult.8,11,12ortho Substitution has been found to slow the thermal relaxation process.10,11 Substitution of all four ortho positions of azobenzene with methoxy groups (compound 1, Scheme 1), or Cl or Br atoms13,14 markedly slows thermal cis to trans relaxation facilitating production of large fractions of the cis isomer.11,14 This substitution pattern also causes separation of the n–π* transitions of cis and trans isomers leading to the possibility of bi-directional visible light photoswitching.13,14 Hecht and colleagues found that tetra-ortho-fluoro substitution also leads to separation of the n–π* transitions of cis and trans isomers without concomitant twisting of the trans geometry, a useful feature in some applications.15 In each of these cases, as well as with C2 bridged azobenzenes,16–18 however, the visible region n–π* transitions are of low intensity. Aprahamian and colleagues recently reported that coordination of the azo group with a BF2 moiety produced promising improvements in the molar absorptivity at visible wavelengths4 (ε512 = 8026 M−1 cm−1). However, this compound is structurally more complex and shows unusual oxygen dependence to its thermal relaxation rate.
 | ||
| Scheme 1 ortho-Substituted azobenzenes studied here. | ||
Several of the slow-relaxing visible-light switching azobenzenes that have been studied (the methoxy-substituted and the C2 bridged compounds) are sensitive to reduction by glutathione at typical intracellular concentrations.13,17,18 This feature limits their use in vivo. The tetra-ortho-chloro compound is stable to glutathione but has low absorptivity.14 Stability to reduction of the tetra-ortho-fluoro compound and the BF2-coordinated azo compound have not been reported.
To address these limitations, we explored replacing ortho oxygen atoms with sulphur atoms. We synthesized compounds 2 and 3 as well as the water soluble versions 4, and 5 (see ESI†). Effects of replacing oxygen with sulphur or higher elements of the chalcogen group on the optical properties of materials have been found to vary from minor19 to dramatic,20 depending on the detailed bonding arrangements and the transitions involved. Thiol substituents in para positions have been reported to induce useful red shifts in azo modified biomolecules by Asanuma and colleagues10 and Kaihatsu and colleagues.9
Fig. 1 shows experimental UV-Vis spectra obtained for 1, 2 and 3. Spectra were also calculated using time-dependent DFT methods implemented in Gaussian 09 at the B3LYP level of theory with the 6-311++g(d,p) basis set and a DMSO solvent model. This treatment has been successful in predicting optical properties of similar azobenzene systems.13,15 Calculated spectra for 1 and for the S-methyl versions of 2 (2′) and 3 (3′) are also shown in Fig. 1. Computations were performed with methyl rather than ethyl versions of the compounds to isolate effects of O-to-S substitution on the spectra.
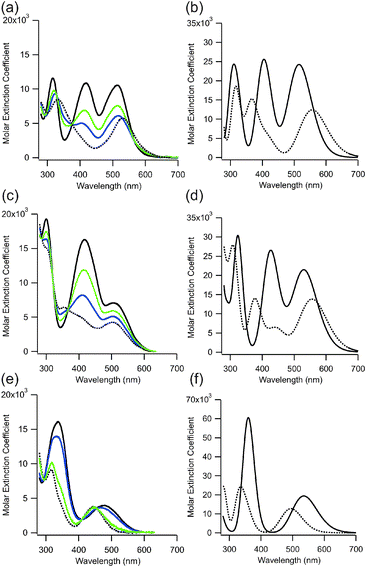 | ||
| Fig. 1 Experimental UV-Vis spectra (panels a, c, e) of 2, 3, and 1, (respectively) in the dark-adapted (E form) in DMSO (solid black line) and with green (530 nm) or blue (450 nm) irradiation. The extrapolated spectrum of the cis isomer is shown as a dotted line (see ESI†). Calculated UV-Vis spectra (panels b, d, f) of 2′, 3′, and 1, (respectively) in the trans (E) form (solid line) and in the cis (Z) form (dotted line) in DMSO. Note the vertical scale in (f) is different. | ||
The experimental data (Fig. 1a, c, and e) show that replacement of oxygen atoms with sulphur at the ortho positions of azobenzenes leads to a >20 nm red shift as well as a significant increase in the intensity of the longest wavelength absorption bands of the trans isomers. Compound 2, in particular, shows ε512 of 10600 M−1 cm−1 in DMSO and compound 3 shows ε505 of 7100 M−1 cm−1 whereas the value for the corresponding oxygen derivative (1) is ε480 = 4030 M−1 cm−1.13
In addition to the enhanced long wavelength absorption, the sulphur containing compounds show strong transitions in the blue region of the spectrum (∼425 nm) (Fig. 1). Calculations indicate these transitions involve HOMO − 1 and HOMO − 2 molecular orbitals and extensive participation of the S atoms (see ESI†). Although differences in the wavelengths of long wavelength (HOMO to LUMO) transitions between trans and cis isomers of 2 and 3 are predicted (Fig. S1, ESI†), experimentally these are not found to be large and instead it is mainly the intensity of the transition that decreases upon trans-to-cis isomerization (Fig. 1). Overall the result of O-to-S substitution is that the sulphur containing photoswitches 2 and 3 undergo trans to cis isomerization with both blue and green light, whereas 1 undergoes trans to cis isomerization with green light and cis to trans isomerization with blue light.13 The photostationary states observed for 2 and 3 depend on the precise irradiation wavelength employed but is limited to ∼75% cis by overlap of the cis and trans spectra (see ESI†) (Fig. 1). This value compares favourably with other visible light switching azobenzenes including 2,2′-ortho substituted and push–pull derivatives.8–10,12,21
In marked contrast to 1, where thermal relaxation of the cis isomer to the thermodynamically more stable trans isomer occurs with a half-life of ∼14 days at 25 °C in DMSO,13 the thermal relaxation of 2 and 3 occurs on a time frame of minutes under the same conditions. From computations, the free energy of the trans forms of 2 and 3 are 0.75 eV and 0.73 eV lower than the cis form, however for 1 the trans form is only 0.33 eV lower in energy than the cis (see ESI†). The greater relative stability of the trans isomer of the S-containing compound presumably results in a lower lying transition state for the thermal cis-to-trans isomerization process. While complete bidirectional switching with no thermal reversion is the ideal case,16,17 a consequence of thermal relaxation on the minutes timescale is that significant fractions of the cis isomer can be produced using moderate light intensities while the trans isomer can be completely repopulated on biologically useful timeframes in the dark. The result is that the extent of photoswitching (i.e. the % change is cis isomer) is large despite overlap of the trans and cis spectra. In addition, the large energy difference between trans and cis isomers can be used to drive conformational changes in attached target molecules.22,23 A relaxation time frame of minutes is well suited for photo-control of transcription factors in vivo, for example.24
The enhanced absorption of these compounds in the visible region of the spectrum and their thermal relaxation on the time frame of minutes provides a significant improvement in terms of biological photoswitching. Essential to their use in intracellular environments in biological settings however, is their stability to reduction by glutathione.
Our analysis of the mechanism of reduction of 1 by glutathione, indicated that the thiol attacks at one nitrogen atom of the azo unit facilitated by prior, or concerted protonation of the other nitrogen atom.14 The azonium ion of 1 is stabilized by resonance delocalization of the methoxy groups as well as H-bonding of the azonium proton to a methoxy oxygen atom.14 Replacement of methoxy groups with S-ethyl groups is expected to decrease stabilization of the azonium ion via resonance delocalization; the Hammett σp constants for –OCH3 and –SCH3 are −0.27 and 0.00 respectively.25 In addition H-bonding to sulphur is expected to be weaker and to have different geometrical preferences, than H-bonding to oxygen.26–28 As a consequence the thiol containing photoswitch should be less sensitive to reduction by glutathione than the oxygen containing counterpart.
To test stability of the thiol-containing photoswitch to reduction by glutathione in a biologically relevant environment, we prepared the water soluble versions 4 and 5. Spectra of these compounds are qualitatively similar in water and in DMSO and similar to those of 2 and 3 (see ESI†). Fig. 2 shows the diacetamido derivative 4 in aqueous phosphate buffer at 20 °C. The diacetamido functional group provides a means for attaching both ends of this photoswitch to a target biomolecule as has been done for a range of related photoswitches.22,23
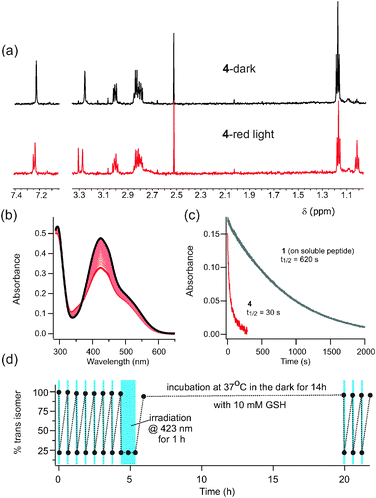 | ||
| Fig. 2 (a) NMR spectra of 4 in D2O after dark adaptation and after exposure to red light (635 nm). (b) UV-Vis spectrum of 4 after dark adaptation (black line) and after irradiation with red light (thick red line). Intermediate scans show thermal relaxation back to the dark state (τ½ = 25 min at 20 °C). (c) Time course for trans to cis isomerization under red light is ∼20 fold faster for 4 than for 1. (1 is attached to FK-11 polyG14 to enhance its water solubility.) (d) Absorbance measured at points indicated over multiple rounds of switching using blue light (423 nm, indicated with blue boxes) with intervening periods of dark adaptation. There is no evidence of photobleaching or reduction by 10 mM reduced glutathione. | ||
Interestingly, the long wavelength absorption tail of the trans isomer of 4 (but not of 5, see ESI†) makes it possible to use red light (635 nm) (Fig. 2a) as well as other visible wavelengths (Fig. 2d) for photoisomerization. The use of red light for isomerization is particularly appealing for biological systems since red wavelengths are much more penetrating through biological tissue than other colors of the visible spectrum.2 We have observed red-light driven trans-to-cis isomerization for tetra-ortho-methoxy and tetra-ortho-chloro species,14 but the enhanced red absorption of 4 leads to significantly faster production of the cis isomer (Fig. 2c). Thermal relaxation of 4 occurs with a half-life of 25 min at 20 °C in aqueous solution (Fig. 2b). The alkylamino derivative 5, shows even faster thermal relaxation (seconds) (see ESI†) and it (or a di-alkylated version) may be useful where faster thermal resetting is required.
Critically, compounds 4 and 5 are not reduced by 10 mM reduced glutathione, conditions designed to mimic intracellular in vivo conditions. This feature is in marked contrast to the tetra-ortho-methoxy compound 1 which is reduced on the time frame of ∼1 h under these conditions.13Fig. 2d shows that photoswitching is preserved after 14 h at 37 °C with 10 mM glutathione. The alkyl substituted compound 5 is also stable to reduction (see ESI†). In addition, there is no evidence of photobleaching of these compounds (Fig. S3c, ESI†). The stability of these ortho-S-alkyl substituted compounds means that a variety of intracellular applications may now be envisaged.
Notes and references
- Molecular switches, ed. B. Feringa and W. R. Browne, Wiley-VCH, 2nd edn, 2011 Search PubMed.
- W. F. Cheong, S. A. Prahl and A. J. Welch, IEEE J. Quantum Electron., 1990, 26, 2166–2185 CrossRef.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 2–34 Search PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2012, 134, 15221–15224 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- D. Bleger, Z. Yu and S. Hecht, Chem. Commun., 2011, 47, 12260–12266 RSC.
- H. A. Wegner, Angew. Chem., Int. Ed., 2012, 51, 4787–4788 CrossRef CAS PubMed.
- A. Mourot, M. A. Kienzler, M. R. Banghart, T. Fehrentz, F. M. E. Huber, M. Stein, R. H. Kramer and D. Trauner, ACS Chem. Neurosci., 2011, 2, 536–543 CrossRef CAS PubMed.
- S. Sawada, N. Kato and K. Kaihatsu, Curr. Pharm. Biotechnol., 2012, 13, 2642–2648 CAS.
- H. Nishioka, X. G. Liang, T. Kato and H. Asanuma, Angew. Chem., Int. Ed., 2012, 51, 1165–1168 CrossRef CAS PubMed.
- O. Sadovski, A. A. Beharry, F. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484–1486 CrossRef CAS PubMed.
- L. Chi, O. Sadovski and G. A. Woolley, Bioconjugate Chem., 2006, 17, 670–676 CrossRef CAS PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- D. Bleger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Nather, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- S. Samanta, C. G. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51, 6452–6455 CrossRef CAS PubMed.
- H. Sell, C. Nather and R. Herges, Beilstein J. Org. Chem., 2013, 9, 1–7 CrossRef CAS PubMed.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef CAS PubMed.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 539–547 CrossRef CAS PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, Org. Biomol. Chem., 2008, 6, 4323–4332 CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- T. Fehrentz, M. Schonberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156–12182 CrossRef CAS PubMed.
- F. Zhang, K. A. Timm, K. M. Arndt and G. A. Woolley, Angew. Chem., Int. Ed., 2010, 49, 3943–3946 CrossRef CAS PubMed.
- C. Hansch and A. Leo, Substituent constants for correlation analysis in chemistry and biology, John Wiley & Sons, New York, 1979 Search PubMed.
- F. H. Allen, C. M. Bird, R. S. Rowland and P. R. Raithby, Acta Crystallogr., Sect. B, 1997, 53, 696–701 CrossRef.
- J. A. Platts, S. T. Howard and B. R. F. Bracke, J. Am. Chem. Soc., 1996, 118, 2726–2733 CrossRef CAS.
- F. Wennmohs, V. Staemmler and M. Schindler, J. Chem. Phys., 2003, 119, 3208–3218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, synthetic procedures, NMR data and further UV/Vis data. See DOI: 10.1039/c3cc46045b |
| This journal is © The Royal Society of Chemistry 2013 |