 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic modification of salinomycin: selective O-acylation and biological evaluation†
Björn Borgströma, Xiaoli Huangb, Martin Poštaa, Cecilia Hegardtc, Stina Oredssonb and Daniel Strand*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, Box 124, 221 00 Lund, Sweden. E-mail: daniel.strand@chem.lu.se; Fax: +46 (0)46 222 82 09; Tel: +46 (0)46 222 81 23
bDepartment of Biology, Lund University, Sweden
cDepartment of Oncology, Clinical Sciences, Lund University, Sweden
First published on 3rd September 2013
Abstract
Salinomycin has found renewed interest as an agent for prevention of cancer recurrence through selectively targeting cancer stem cells. Strategies for generation of improved salinomycin analogs by individual modification of its hydroxyl groups are presented. An evaluation of the dose–response effects of the resulting library on breast cancer cell lines shows that acylation of the C20 hydroxyl can be used to improve IC50 values down to one fifth that of salinomycin.
Cancer stem cells (CSCs) are a subpopulation of cancer cells that have been invoked in recurrence, multi-drug resistance, and metastasis of cancer.1 Some of these effects are likely related to a reduced sensitivity to conventional chemotherapy in these cells.1a Small molecules that selectively target the CSC population, either by inducing a phenotypical change or by inhibiting growth, thus constitute an important new avenue for preventing recurrence and metastasis of cancer as most current therapies do not address stem-like cancer cells. Methods to identify such compounds by high-throughput screening have been developed.2 Salinomycin (SA, 1)3,4 was recently identified as the most selective in a library of more than 16
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds in reducing the proportion of putative CSCs in breast cancer cell lines as well as inhibiting mammary tumor growth in vivo.2 The mechanism by which SA influences the CSC population is not fully understood.5a,b Proposed modes of action include inhibition of P-glycoprotein gp170,5c interference of the Wnt signaling cascade,5d increased DNA damage and reduction of the protein p21 level,5e overcoming ABC transporter-mediated multidrug and apoptosis resistance,5a increasing oxidative stress,5f and increasing levels of reactive oxygen species.5g,h The recent surge of activity related to SA in the biomedical field has yet to transgress to the synthetic community; a report of C1 amides of SA being the exception.6 Semi synthesis of analogs through selective chemical modification of SA constitutes an attractive avenue for identifying compounds with improved selectivity against CSCs and equally important, for advancing the structural understanding of the mode of action of SA against them. In particular, perturbation of the ion transport properties of SA by systematic manipulation of the substructures involved in ion binding is of value to this end as the antiporter properties of SA has been suggested as an origin for its CSC activity.7 Herein, we describe general and selective synthetic strategies for the individual modification of all hydroxyl groups in SA,8 as well as an evaluation of the resulting analog library in breast cancer cells. Significantly, SA analogs acylated at the C20 hydroxyl displayed IC50 values down to one fifth that of the native structure against breast cancer cells in an MTT-based dose–response assay. The synthetic procedures described are of value for the development of improved SA derivatives as well as for the exploration of SAR for its CSC selectivity.9
000 compounds in reducing the proportion of putative CSCs in breast cancer cell lines as well as inhibiting mammary tumor growth in vivo.2 The mechanism by which SA influences the CSC population is not fully understood.5a,b Proposed modes of action include inhibition of P-glycoprotein gp170,5c interference of the Wnt signaling cascade,5d increased DNA damage and reduction of the protein p21 level,5e overcoming ABC transporter-mediated multidrug and apoptosis resistance,5a increasing oxidative stress,5f and increasing levels of reactive oxygen species.5g,h The recent surge of activity related to SA in the biomedical field has yet to transgress to the synthetic community; a report of C1 amides of SA being the exception.6 Semi synthesis of analogs through selective chemical modification of SA constitutes an attractive avenue for identifying compounds with improved selectivity against CSCs and equally important, for advancing the structural understanding of the mode of action of SA against them. In particular, perturbation of the ion transport properties of SA by systematic manipulation of the substructures involved in ion binding is of value to this end as the antiporter properties of SA has been suggested as an origin for its CSC activity.7 Herein, we describe general and selective synthetic strategies for the individual modification of all hydroxyl groups in SA,8 as well as an evaluation of the resulting analog library in breast cancer cells. Significantly, SA analogs acylated at the C20 hydroxyl displayed IC50 values down to one fifth that of the native structure against breast cancer cells in an MTT-based dose–response assay. The synthetic procedures described are of value for the development of improved SA derivatives as well as for the exploration of SAR for its CSC selectivity.9Pioneering work detailed the use of aliphatic acid anhydrides to esterify the C20 hydroxyl group.3b,10 General access to such structures would be of particular value in a CSC context as such modifications resulted in an enhanced ion binding and activity in antibiotic assays.3 In our hands, this method was however limited to unhindered acid anhydrides and we were only able to isolate a subset of such structures in homogenous form in low yields. Towards circumventing these problems, reversible masking of the carboxylic acid of SA is a key enabling modification. Protection of this moiety enhances the stability of intermediates and facilitates both purification and characterization of intermediates in analog synthesis. Conversion of SA into the known methyl ester 3a was accomplished in good yield with TMSCHN2 (Scheme 1). Attempts to hydrolyze this ester under basic conditions however proved to be unsuccessful. Also milder protocols including the use of Me3SnOH11 resulted in decomposition of the sensitive structure. A suitable protective group for the carboxylate, TMSEt-, could however be introduced in 61% yield, also on a multi-gram scale, using TCFH12 as the coupling reagent. This result is noteworthy in light of the comparatively low nucleophilicity of TMSEtOH and the steric inaccessibility of the acid; the difficulty of using coupling reagents with SA has been commented on13 and a range of conventional coupling reagents (DCC, EDC/HOBt, HATU, PyBOP etc.) failed to give even trace amounts of ester 3b. Importantly, SA could be quantitatively and tracelessly released from the TMSEt-ester 3b with TBAF in THF. Washing the crude product with Na2CO3 (aq.) gave the SA·Na salt (2), indistinguishable from the natural product, and pure by 1H- and 13C-NMR spectroscopy. For an orthogonal alternative to TBAF deprotection, allyl ester 3c was also prepared by treatment of 2 with allyl bromide in the presence of Cs2CO3. Cleavage of this ester with Pd(0) returned SA in moderate yield (41%).
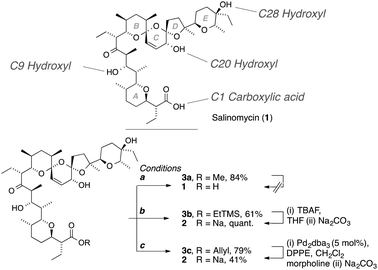 | ||
| Scheme 1 Alkylation and deprotection of the carboxylic acid of SA. Conditions (a) TMSCHN2, toluene–MeOH 2:1; (b) TMSEtOH, TCFH, DIPEA, CH2Cl2, 0°; (c) Cs2CO3, allyl bromide, DMF. | ||
In contrast to the SA or the SA·Na salt, protected 3b can be reacted cleanly and selectively with a variety of acid chlorides at the C20 hydroxyl (Scheme 2). Reactions of unprotected SA (or its Na salt) with BzCl, DMAP, and Et3N or Bz2O gave either no reaction or extensive side product formation. We were unable to isolate benzoate 5b from any of these experiments. The TMSEt-esters of 4a–q could all be cleanly cleaved with TBAF and the products isolated as their sodium salts. In addition, C20 carbonates were readily formed from reactions with chloroformates. A C20-carbamoylated product was formed from the reaction with PhNCO. Less reactive isocyanates (Et- and tBu-) required addition of catalytic amounts of CuCl to react.14 After deprotection, carbamates and carbonates 5m–5q were isolated in good yields over three steps. To enable selective derivatization of the C9 and C28 alcohols, silyl groups were introduced (Scheme 3). Selective silylation of the C20 hydroxyl was accomplished quantitatively with TESCl and imidazole in CH2Cl2. Somewhat surprisingly, the most reactive alcohol of 6 was the tertiary alcohol at C28. Treatment of 6 with an excess of PhNCO and CuCl in DMF carbamoylated this position selectively. Excess EtNCO with CuCl gave allophenate 9c under the same conditions. By limiting the amount of isocyanate, ethyl carbamate 9b could also be isolated in low conversion. Removal of the protective groups with TBAF gave carbamates 9a/b and allophenate 9c in good yields over two steps. The position of the acyl groups at C28 was confirmed by diagnostic downfield shifts of the C28 signal in the respective 13C-NMR spectra (15.6–10.2 ppm).15 Towards modifying the least reactive C9 hydroxyl, bis-silylation of both the C20 and C28 alcohols of 3b/c was accomplished with TESCl in DMF. Minor amounts of tri-TES side-product were also isolated under these conditions. Global deprotection of tri-TES 8 with TBAF corroborated the retained stereochemical integrity of this compound; all four protective groups were cleanly removed and the SA·Na salt was obtained as a single diastereomer indistinguishable from the natural product by 1H-NMR.
 | ||
| Scheme 2 Selective acylation at the 20-position. Conditions (a): RCOCl, DMAP, Et3N, CH2Cl2 (5a–5l); (b) RNCO, CH2Cl2 (5m); (c) RNCO, CuCl, DMF (5n, 5o); (d) ROCOCl, DMAP, Et3N, CH2Cl2 (5p and 5q). aIsolated yields over three steps. | ||
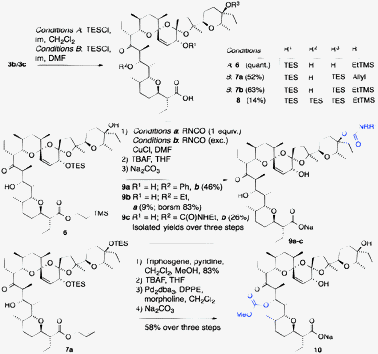 | ||
| Scheme 3 Acylation of the C9 and C28 hydroxyls. | ||
Acylation of the C9 alcohol was accomplished by reacting bis-TES 7a/b with triphosgene followed by addition of methanol to give the corresponding methyl carbonates. Removal of the TMSEt-group with TBAF however proved to be incompatible with this modification due to competing elimination of the carbonate.16 Instead, the use of allyl ester 7a provided a milder alternative; removal of the TES-groups by brief exposure to TBAF, followed by release of the carboxylic acid under Pd(0) catalysis gave carbonate 10 in 58% overall yield. The position of the carbonate at C9 was confirmed by a diagnostic HMBC correlation from the C9 proton to the carbonate carbonyl carbon.15
The antiproliferative activity of the O-acylated analogs and methyl ester 3a was evaluated in MCF-7 and JIMT-1 breast cancer cells using an MTT assay (Table 1). The C20-acylated analogs displayed IC50 values lower or similar to that of SA in both cell lines. The enhancement of activity in antibiotic assays for C20 esters is thus carried over to cancer cells. The most potent C20 analogs in this assay proved to be those with the least bulky substituents in each series, ethyl carbonate 5q, acetate 5g, and ethyl carbamate 5n. Analogs deprived of a stabilizing interaction between the carboxylate and the C9 hydroxyl (3a and 10) exhibited a significantly reduced activity. The retained activity of the C28 carbamates 9a/b is in line with the previous suggestion that in membranes, unlike in the solid state, this group does not contribute to the ion-binding of SA by hydrogen bonding to the carboxylate.17

In summary, acylation of the C20 hydroxyl groups of the therapeutically promising compound SA gave analogs with IC50 values down to one fifth that of the native structure against cancer cells. Selective introduction of protective groups enables diverse access to analogs individually modified at the C9, C20, and C28 hydroxyl groups. Certain acyl derivatives of the C28 hydroxyl retain the activity of the parent compound whereas derivatization of positions directly involved in ion-binding reflects in a reduced activity. The strategies presented should be of significant value also for more elaborate modifications of SA. An extensive biological investigation of the CSC selectivity of these and related compounds and the correlation of this selectivity with antiporter activity is currently underway and will be reported in due course.
The Crafoord Foundation, the Magn Bergvall Foundation, The Royal Academy of Sciences, the Johan Hierta Foundation, the Mrs Berta Kamprad Foundation, the Percy Falk Foundation, and The Swedish Research Council are acknowledged for funding. We thank Prof. Lo Persson for insightful discussions.
Notes and references
- For lead references, see: (a) M. Dean, T. Fojo and S. Bates, Nat. Rev. Cancer, 2005, 5, 275 CrossRef CAS PubMed; (b) N. Y. Frank, T. Schatton and M. H. Frank, J. Clin. Invest., 2010, 120, 41 CrossRef CAS PubMed; (c) K. Sampieri and R. Fodde, Semin. Cancer Biol., 2012, 22, 187 CrossRef CAS PubMed; (d) For recent reviews, see: C. Naujokat and R. Steinhart, J. Biomed. Biotechnol., 2012, 2012, 950658 CrossRef PubMed.
- P. B. Gupta, T. T. Onder, G. Jiang, K. Tao, C. Kuperwasser, R. A. Weinberg and E. S. Lander, Cell, 2009, 138, 645 CrossRef CAS PubMed.
- (a) Y. Miyazaki, M. Shibuya, H. Sugawara, O. Kawaguchi and C. Hirsoe, J. Antibiot., 1974, 27, 814 CrossRef CAS; (b) Y. Miyazaki, H. Kinashi, N. Otake, M. Mitani and T. Yamanishi, Agric. Biol. Chem., 1976, 40, 1633 CrossRef CAS.
- For total synthesis of SA, see: (a) Y. Kishi, S. Hatakeyama and M. D. Lewis, in Frontiers of Chemistry, ed. K. J. Laidler, Oxford, 1982 Search PubMed; (b) K. Horita, S. Nagato, Y. Oikawa and O. Yonemitsu, Chem. Pharm. Bull., 1989, 37, 1698 CrossRef CAS; (c) P. J. Kocienski, R. C. D. Brown, A. Pommier, M. Procter and B. Schmidt, J. Chem. Soc., Perkin Trans. 1, 1998, 9 RSC.
- (a) A. Huczyński, Chem. Biol. Drug Des., 2012, 79, 235 CrossRef PubMed; (b) A. Huczyński, Bioorg. Med. Chem. Lett., 2012, 22, 7002 CrossRef PubMed; (c) R. Riccioni, M. L. Dupuis, M. Bernabei, E. Petrucci, L. Pasquini, G. Mariani, M. Cianfriglia and U. Testa, Blood Cells, Mol., Dis., 2010, 45, 86 CrossRef CAS PubMed; (d) D. S. Lu, M. Y. Choi, J. Yu, J. E. Castro, T. J. Kipps and D. A. Carson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13253 CrossRef CAS PubMed; (e) J.-H. Kim, M. Chae, W. K. Kim, Y.-J. Kim, H. S. Kang, H. S. Kim and S. Yoon, Br. J. Pharmacol., 2011, 162, 773 CrossRef CAS PubMed; (f) K. Ketola, M. Hilvo, T. Hyötyläinen, A. Vuoristo, A.-L. Ruskeepää, M. Orešič, O. Kallioniemi and K. Iljin, Br. J. Cancer, 2012, 106, 99 CrossRef CAS PubMed; (g) B. Verdoodt, M. Vogt, I. Schmitz, S.-T. Liffers, A. Tannapfel and A. Mirmohammadsadegh, PLoS ONE, 2012, 7, e44132 CAS; (h) K.-Y. Kim, S.-N. Yu, S.-Y. Lee, S.-S. Chun, Y.-L. Choi, Y.-M. Park, C. S. Song, B. Chatterjee and S.-C. Ahn, Biochem. Biophys. Res. Commun., 2011, 413, 80 CrossRef CAS PubMed.
- (a) A. Huczyński, J. Janczak, M. Antoszczak, J. Wietrzyk, E. Maj and B. Brzezinski, Bioorg. Med. Chem. Lett., 2012, 22, 7146 CrossRef PubMed; (b) A. Huczyński, J. Janczak, J. Stefanska, M. Antoszczak and B. Brzezinski, Bioorg. Med. Chem. Lett., 2012, 22, 4697 CrossRef PubMed.
- J. Tulla-Puche, Á. Torres, P. Calvo, M. Royo and F. Albericio, Bioconjugate Chem., 2010, 3, 555 Search PubMed.
- For examples of selective derivatization of hydroxyl groups in complex natural products, see: (a) J. González-Sabín, R. Morán-Ramallal and F. Rebolledo, Chem. Soc. Rev., 2011, 40, 5321 RSC; (b) R. Tong and J. J. Cheng, Chem. Sci., 2012, 3, 2234 RSC.
- For the SAR of the antibiotic salinomycin, see ref. 3b.
- P. Hammann, W. Raether and L. Vertesy, J. Antibiot., 1993, 46, 523 CrossRef CAS.
- K. C. Nicolaou, A. A. Estrada, M. Zak, S. H. Lee and B. S. Safina, Angew. Chem., Int. Ed., 2005, 44, 1378 CrossRef CAS PubMed.
- J. Tulla-Puche, Á. Torres, P. Calvo, M. Royo and F. Albericio, Bioconjugate Chem., 2008, 19, 1968 CrossRef CAS PubMed.
- A. Huczyński, J. Janczak, M. Antoszczak, J. Stefańska and B. Brzezinski, J. Mol. Struct., 2012, 1002, 197 CrossRef PubMed.
- M. E. Duggan and J. S. Imagire, Synthesis, 1989, 131 CrossRef CAS.
- The full assignment was based on a combination of COSY, HMQC, and HMBC correlations. See ESI† for details.
- HF·pyr in THF gave clean deprotection of the C20 O-TES group of 7a. Other fluoride sources (HF (aq), HF·Et3N, and CsF) and solvents were screened but either led to elimination at C9, partial deprotection, or decomposition by cleavage across the C-ring, see; J. L. Wells, J. Bordner, P. Bowles and J. W. McFarland, J. Med. Chem., 1988, 31, 274 CrossRef CAS.
- (a) E. F. Paulus, M. Kurz, H. Matter and L. Vertesy, J. Am. Chem. Soc., 1998, 120, 8209 CrossRef CAS; (b) N. Matsumori, A. Morooka and M. Murata, J. Am. Chem. Soc., 2007, 129, 14989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra of all new compounds and MTT dose–response curves. See DOI: 10.1039/c3cc45983g |
| This journal is © The Royal Society of Chemistry 2013 |