 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triplex-mediated analysis of cytosine methylation at CpA sites in DNA†
Marie W.
Johannsen‡
a,
Simon R.
Gerrard
a,
Tracy
Melvin
b and
Tom
Brown‡
*a
aSchool of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: tom.brown@chem.ox.ac.uk; Tel: +44 (0)1865 275413
bOptoelectronics Research Centre, University of Southampton, SO17 1BJ, UK
First published on 7th November 2013
Abstract
Modified triplex-forming oligonucleotides distinguish 5-methyl cytosine from unmethylated cytosine in DNA duplexes by differences in triplex melting temperatures. The discrimination is sequence-specific; dramatic differences in stabilisation are seen for CpA methylation, whereas CpG methylation is not detected. This direct detection of DNA methylation constitutes a new approach for epigenetic analysis.
The existence of 5-methyl cytosine (MeC) in genomic DNA has been known for more than 60 years.1 Although it has been established as an epigenetic marker involved in gene expression and transposable element suppression in mammals, plants and fungi, its full range of functions are still not fully understood.2 In mammals cytosine methylation is estimated to occur at 70–80% of CpG sites in the genome.3 Non-CpG methylation also occurs, most notably in mammalian embryonic stem cells (ESCs),4 plants and insects.3,5 Higher relative levels of methylation of CpA sites are observed in pluripotent ESCs compared to differentiated cells, so CpA methylation is suggested to play a role in the origin and maintenance of the pluripotent lineage.4
In order to develop a complete understanding of the biological consequences of cytosine methylation in DNA, sequence-specific detection methods for MeC are urgently required. Several indirect methods exist,6 the most widespread being bisulphite sequencing. Treatment of DNA with bisulphite converts C to U, leaving MeC unchanged; subsequent sequencing of the treated and untreated samples allows for differentiation between C and MeC.7 However, this method is labour-intensive, much of the DNA is destroyed in the process, and incomplete conversion of C to U leads to sequencing errors.2c,6,7b Direct detection methods have been investigated,8 but are not in general use.
With a view to providing sequence dependent detection of DNA methylation we investigated the potential of triplex formation to discriminate MeC from C. A series of modified triplex-forming oligonucleotides (TFOs) were previously shown by us to yield stable triple helices at neutral pH with duplexes containing all four Watson–Crick base pair combinations (AT, TA, GC, CG).9 In these studies several analogues of N-methylpyrrolo-dC were investigated for improved CG recognition (Fig. 1).10 It was noted that when the modified nucleotide XP was placed opposite to MeCG instead of CG, the thermal stability of the triplex was greatly decreased. This is illustrated in the denaturation curves in Fig. 2.
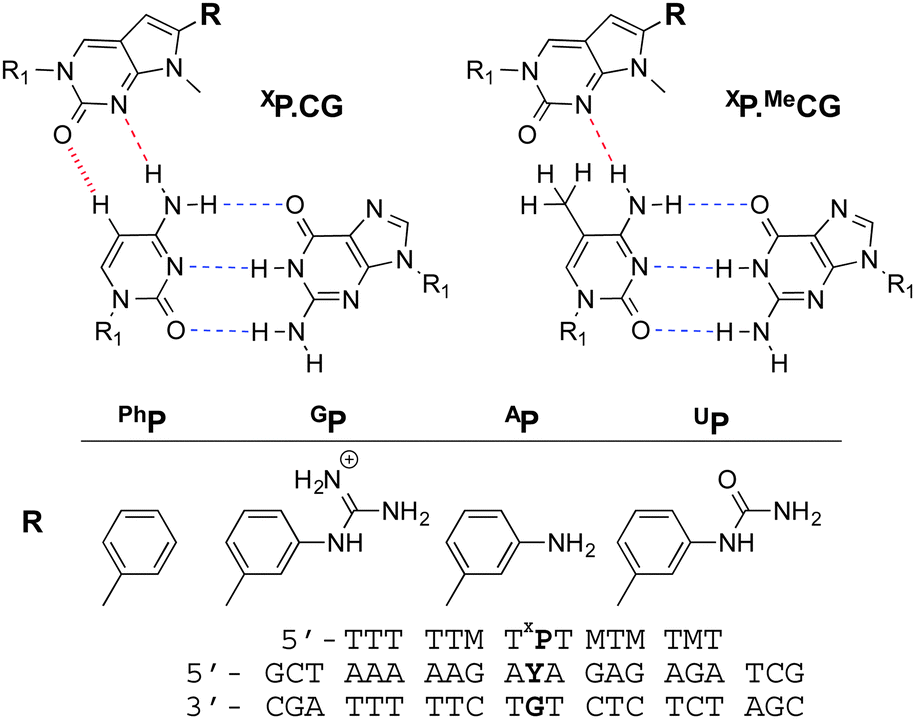 | ||
| Fig. 1 Proposed structure for the XP.CG and XP.MeCG triplets. H-bonds are shown with dashed lines, while for C, a potential dipolar C–H⋯O interaction is shown with a hashed line. R1 = 2′-deoxyribofuranose. Also shown are the modified nucleotides incorporated in the triplex forming oligonucleotide sequence and the DNA duplex (set 1) studied. M = MeC, Y = C or MeC. | ||
 | ||
Fig. 2 Thermal denaturation of set 1 triplexes with PhP. (A) UV absorption at 260 nm recorded as a function of temperature from 15 °C to 80 °C. (B) Smoothed first derivative of the thermal denaturation curves shown in A. The first transition represents triplex denaturation (TFO dissociation), the second is denaturation of the duplex. 10 mM phosphate buffer, 200 mM NaCl, 1 mM EDTA, pH 6.2. PhP.CG (—), PhP.MeCG ( ). ). | ||
The incorporation of PhP into the TFO opposite a CG or MeCG base pair in the duplex provided the largest difference in triplex denaturation temperature relative to other XP monomers. Replacement of PhP by thymine (X = T) gave no discrimination between MeC and C (Table 1 and Table S1†).
| T | PhP | AP | UP | GP | |
|---|---|---|---|---|---|
| T m data measured in °C. All experiments performed with oligonucleotide set 1. 10 mM phosphate buffer, 200 mM NaCl, 1 mM EDTA, pH 6.2. Final Tm values are averages of at least three measurements, except where marked*, which is the average of two measurements. | |||||
| C | 26.7* | 32.0 | 31.5 | 30.9* | 29.0* |
| MeC | 26.7* | 18.1 | 19.2 | 21.8* | 20.2* |
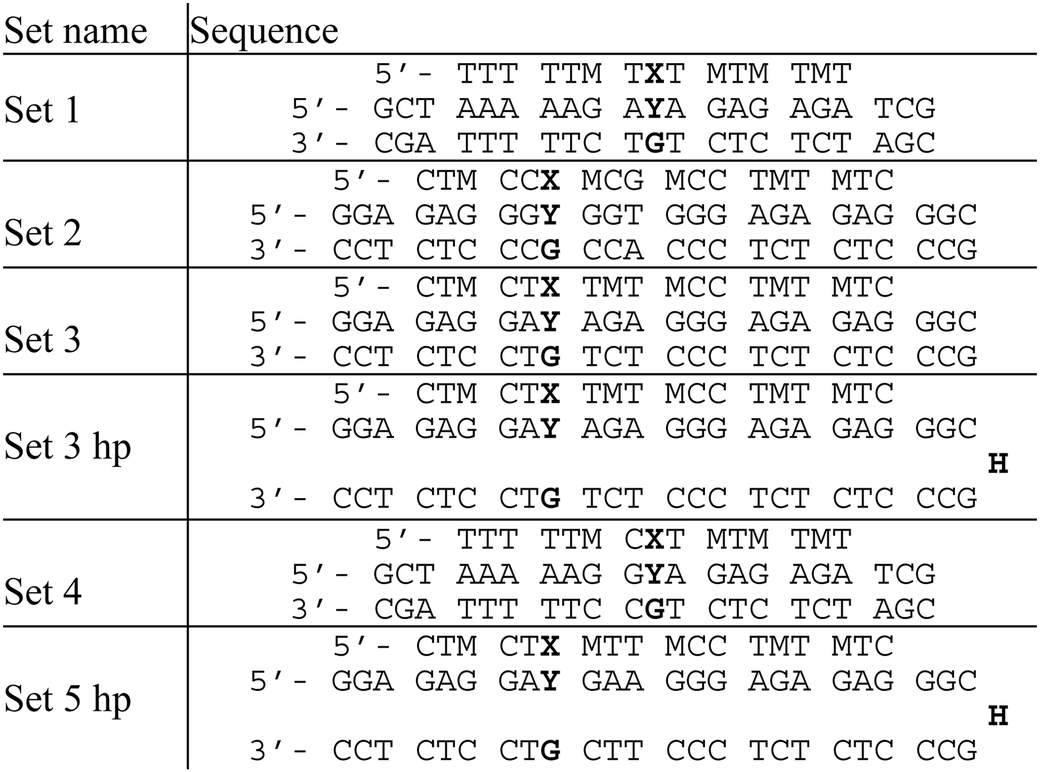
The sequence dependence of triplex melting was next evaluated using TFOs containing PhP and several duplexes containing C and MeC. The epigenetically relevant CpG and CpA dinucleotide sequences were incorporated in some of the DNA duplexes (Table 2). The duplexes in set 3 hp and set 5 hp are hairpins linked via a hexaethylene glycol spacer.
| M = MeC, X = PhP, Y = C or MeC, H = hexaethylene glycol. |
|---|
|
The melting temperatures of the DNA duplexes (Table 2) containing MeC and C flanked by adenine or guanine are shown in Table 3. The differences in denaturation temperature (ΔTm) between equivalent MeC and C duplexes followed the trend AYA > GYA > AYG > GYG. Triplex discrimination of DNA sequences containing CpA is particularly striking. For the GYG containing duplex (set 2) a much lower triplex melting temperature was observed irrespective of the incorporation of MeC or C in the duplex DNA. This is due to the presence of a TA inversion (G.TA triplet) in the triplex.
| Set name | Target trinucleotide | C | MeC | ΔTm |
|---|---|---|---|---|
| T m data measured in °C. Y = C or MeC. 10 mM phosphate buffer, 200 mM NaCl, 1 mM EDTA, pH 6.2. Final Tm values are averages of at least three measurements.a A transition is also seen at lower temperature, most likely due to weak duplex secondary structure. It is also observed when the Tm is measured for the duplex only, and is not seen in the hairpin structure (Tables S2 and S3†). | ||||
| Set 1 | AYA | 32.0 | 18.1 | 13.9 |
| Set 2 | GYG | 16.5 | 16.7 | −0.2 |
| Set 3 | AYA | 32.4a | 21.6 | 10.8 |
| Set 3 hp | AYA | 32.1 | 22.9 | 9.2 |
| Set 4 | GYA | 25.5 | 17.8 | 7.8 |
| Set 5 hp | AYG | 27.0 | 24.1 | 2.9 |
The triplex stability of set 2 was also investigated with X = T in place of PhP. As expected, no significant difference was seen in the Tm values (Tm = 16.9 °C when Y = C, Tm = 17.0 °C when Y = MeC). Set 3 and set 3 hp are identical in sequence (Table 2), and show little difference in triplex Tm, indicating that hairpin and two-stranded duplexes have very similar thermal properties. C/MeC discrimination in different triple helices with the same central tri- and dinucleotide sets is not quite identical, but the similarities between sequences with identical central motifs far outweigh the influence of the wider sequence context.
The same melting studies were performed at pH 5.8, and as expected the triplex melting temperatures were slightly higher compared to those at pH 6.2 (Table S5†). This is because at pH 5.8, the cytosine base will be protonated, leading to stabilisation of the triplex compared with higher pH values.11 The lower pH did not impact significantly upon triplex discrimination of C and MeC containing DNA duplexes. The guanidinylated pyrrolopyrimidine analogue10GP was then evaluated at pH 5.8 in set 2 and set 5 to determine if this would show better discrimination towards MeCpG. This was not the case and GP was not investigated further (Table S1†).
Other epigenetic modifications of importance include 5-hydroxymethylcytosine (hmC), 5-formylcytosine (fC), and 5-carboxycytosine (caC). These are considered to be either intermediates in cytosine demethylation or epigenetic marks in their own right.12 To test the ability of triplex probes to differentiate between these cytosine modifications, the interactions between DNA duplexes (sets 1 and 2, Table 2) and TFOs containing hmC, fC and caC were evaluated at pH 5.8 (Table 4).
| Set name | Target trinucleotide | C | MeC | hmC | fC | caC |
|---|---|---|---|---|---|---|
| T m data measured in °C. Y = C, MeC, hmC, fC or caC as written. 10 mM phosphate buffer, 200 mM NaCl, 1 mM EDTA, pH 5.8. Final Tm values are averages of at least three measurements. | ||||||
| Set 1 | AYA | 40.0 | 25.1 | 24.4 | 29.3 | 27.1 |
| Set 2 | GYG | 23.2 | 23.0 | 22.4 | 23.3 | 22.9 |
For set 1, caC and especially fC were found to afford triplexes of higher thermal stability than MeC and hmC, though still lower than the Tm found for C. Although these differences are modest, there is promise that the triplex approach could be used to distinguish fC from the other C-analogues. The triplex melting for set 2 was the same irrespective of the cytosine modification incorporated in the duplex.
The key observation from the above studies is that TFOs containing PhP opposite CG base pairs at CpA sites strongly stabilise triplexes, but produce very unstable triplexes if the C is replaced by MeC, hmC, and to a lesser extent fC and caC. The structural basis of this has not yet been elucidated, and although the triplex structures in Fig. 1 explain the difference in stability between C and MeC triplexes, they do not explain the observed sequence dependence of triplex melting. This may be influenced by the stability of the triplex region immediately surrounding PhP in addition to other factors. It is noteworthy that 5-methylation of C enhances hydrophobic interactions and has been found to influence DNA intercalation.13 Intercalation of PhP in addition to, or instead of hydrogen-bonding (Fig. 1) cannot be ruled out, in which case the intercalation energy could be influenced by methylation of the duplex. It is also known that CpA methylation affects duplex structure in a manner distinct from CpG methylation. While CpG methylation appears to increase the rigidity of DNA, methylation at CpA sites appears to cause local conformational changes, and an overall increase in the curvature of DNA.14 It is possible that this structural change is responsible for the remarkable difference in stabilisation seen with PhP in the different triplex sequence contexts.
It is surprising that CpA and non-CG methylation in general (CpH methylation) has not previously been the focus of systematic investigation; CpA is the second most common methylation site in most cell types,4,14a,15 and CpA sites are more abundant than CpG sites.16,17 CpH methylation has been linked to silencing of cancer genes in lymphoma and myeloma cell lines,18 and significant levels of CpH methylation have also been found in stably integrated plasmids19 and in human skeletal muscle.20 Interestingly, bisulphite sequencing is thought to underestimate the amount of CpH methylation unless primers are carefully designed to take this into account.20 CpA is thus an interesting and important target for methylation detection.
In conclusion, when the PhP nucleobase analogue is incorporated in TFOs, its effect on triplex stability in certain sequence contexts is strongly dependent on the methylation status of the cytosine directly opposite in the purine strand of the duplex. This method of determining cytosine methylation status has the advantage of being non-destructive towards the analysed DNA and does not require denaturation of double-stranded helices. The findings reported here demonstrate the promise of triplex probes for the determination of methylation status at specific DNA duplex sequences, as well as detection of other epigenetic marks such as fC. Systematic studies are underway on chemically modified TFOs in an attempt to extend the range of cytosine sequences containing epigenetically relevant modifications that can be analysed by this novel approach. Work is also in progress on epigenetically modified systems to enable us to fully understand the observed sequence-dependence of triplex stability.
This research was funded by BBSRC grant BB/I022791: “Detecting cytosine methylation at the single DNA molecule level.” Assistance with oligonucleotide synthesis from ATDBio is gratefully acknowledged.
Notes and references
- (a) G. R. Wyatt, Nature, 1950, 166, 237–238 CrossRef CAS; (b) G. R. Wyatt, Biochem. J., 1951, 48, 581–584 CAS.
- (a) The ENCODE Project Consortium, Nature, 2012, 489, 57–74 CrossRef PubMed; (b) R. E. Thurman, et al. , Nature, 2012, 489, 75–82 CrossRef CAS PubMed; (c) D. D. Deobagkar, C. Panikar, S. N. Rajpathak, N. S. Shaiwale and S. Mukherjee, Methods, 2012, 56, 260–267 CrossRef CAS PubMed.
- J. Arand, D. Spieler, T. Karius, M. R. Branco, D. Meilinger, A. Meissner, T. Jenuwein, G. Xu, H. Leonhardt, V. Wolf and J. Walter, PLoS Genet., 2012, 8, e1002750 CAS.
- (a) R. Lister, et al. , Nature, 2009, 462, 315–322 CrossRef CAS PubMed; (b) B. H. Ramsahoye, D. Biniszkiewicz, F. Lyko, V. Clark, A. P. Bird and R. Jaenisch, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5237–5242 CrossRef CAS.
- (a) E. J. Finnegan, W. J. Peacock and E. S. Dennis, Curr. Opin. Genet. Dev., 2000, 10, 217–223 CrossRef CAS; (b) O. V. Dyachenko, T. V. Shevchuk, L. Kretzner, Y. I. Buryanov and S. S. Smith, Epigenetics, 2010, 5, 569–572 CrossRef CAS.
- (a) C. Piperi, E. Farmaki, F. Vlastos, A. G. Papavassiliou and N. Martinet, J. Biomol. Tech., 2008, 19, 281–284 Search PubMed; (b) P. W. Laird, Nat. Rev. Genet., 2010, 11, 191–203 CrossRef CAS PubMed.
- (a) M. Frommer, L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt, G. W. Grigg, P. L. Molloy and C. L. Pau, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1827–1831 CrossRef CAS; (b) R. P. Darst, C. E. Pardo, L. Ai, K. D. Brown and M. P. Kladde, Curr. Protoc. Mol. Biol., 2010, 91 DOI:10.1002/0471142727.mb0709s91 , 7.9.1-7.9.7.
- J.-L. H. A. Duprey, Z. Zhao, D. M. Bassani, J. Manchester, J. S. Vyle and J. H. R. Tucker, Chem. Commun., 2011, 47, 6629–6631 RSC.
- D. A. Rusling, V. E. C. Powers, R. T. Ranasinghe, Y. Wang, S. D. Osborne, T. Brown and K. R. Fox, Nucleic Acids Res., 2005, 33, 3025–3032 CrossRef CAS PubMed.
- S. R. Gerrard, M. M. Edrees, I. Bouamaied, K. R. Fox and T. Brown, Org. Biomol. Chem., 2010, 8, 5087–5096 CAS.
- J. L. Asensio, A. N. Lane, J. Dhesi, S. Bergqvist and T. Brown, J. Mol. Biol., 1998, 275, 811–822 CrossRef CAS PubMed.
- T. Carell, C. Brandmayr, A. Hienzsch, M. Müller, D. Pearson, V. Reiter, I. Thoma, P. Thumbs and M. Wagner, Angew. Chem., Int. Ed., 2012, 51, 7110–7131 CrossRef CAS PubMed.
- (a) N. Zhang, C. Lin, X. Huang, A. Kolbanovskiy, B. E. Hingerty, S. Amin, S. Broyde, N. E. Geacintov and D. J. Patel, J. Mol. Biol., 2005, 346, 951–965 CrossRef CAS PubMed; (b) J. Bunkenborg, M. M. Stidsen and J. P. Jacobsen, Bioconjugate Chem., 1999, 10, 824–831 CrossRef CAS PubMed.
- (a) Y. Shimooka, J. Nishikawa and T. Ohyama, Biochemistry, 2013, 52, 1344–1353 CrossRef CAS PubMed; (b) Y. Hodges-Garcia and P. J. Hagerman, Biochemistry, 1992, 31, 7595–7599 CrossRef CAS.
- T. Ichiyanagi, K. Ichiyanagi, M. Miyake and H. Sasaki, Nucleic Acids Res., 2013, 41, 738–745 CrossRef CAS PubMed.
- K. Jabbari and G. Bernardi, Gene, 2004, 333, 143–149 CrossRef CAS PubMed.
- D. M. Woodcock, P. J. Crowther and W. P. Diver, Biochem. Biophys. Res. Commun., 1987, 145, 888–894 CrossRef CAS.
- (a) M. C. Lorincz and M. Groudine, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10034–10036 CrossRef CAS PubMed; (b) C. S. Malone, M. D. Miner, J. R. Doerr, J. P. Jackson, S. E. Jacobsen, R. Wall and M. Teitel, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10404–10409 CrossRef CAS PubMed.
- S. J. Clark, J. Harrison and M. Frommer, Nat. Genet., 1995, 10, 20–27 CrossRef CAS PubMed.
- (a) R. Barrès, M. E. Osler, J. Yan, A. Rune, T. Fritz, K. Caidahl, A. Krook and J. R. Zierath, Cell Metab., 2009, 10, 189–198 CrossRef PubMed; (b) J. Yan, J. R. Zierath and R. Barrès, Exp. Cell Res., 2011, 317, 2555–2561 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation of oligonucleotides, additional melting data. See DOI: 10.1039/c3cc45917a |
| ‡ Current address: Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Rd, Oxford, OX1 3TA, UK. |
| This journal is © The Royal Society of Chemistry 2014 |