 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RYB tri-colour electrochromism based on a molecular cobaloxime†
Maik R. J. Scherer‡a, Nicoleta M. Muresan‡b, Ullrich Steiner*a and Erwin Reisner*b
aCavendish Laboratory, Department of Physics, University of Cambridge, J J Thomson Avenue, Cambridge CB3 0HE, UK. E-mail: u.steiner@phy.cam.ac.uk
bChristian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 2nd September 2013
Abstract
The three oxidation states of Co in a molecular cobaloxime were used to realise an electrochromic device displaying the red, yellow, blue (RYB) set of subtractive primary colours. A facile method for the lithographic patterning of a several micrometre thick indium tin oxide (ITO) mesoporous layer was developed, which served as the scaffold for cobaloxime adsorption.
The fabrication of electrochromic devices and displays based on hybrid materials is currently attracting much attention.1 Full colour displays require the superposition of three primary colours, with red-green-blue (RGB) and cyan-yellow-magenta-black (CYMK) being the most common additive and subtractive colour models. A historical alternative is the artist's primary colour wheel containing red, yellow and blue (RYB). Most electrochromic materials however alternate between two colours only and full-colour electrochromic displays are rare and typically based on conjugated polymers.2
Supra-architectural assemblies comprising conjugated conducting polymers such as polythiophene,3 polypyrrole,4 polyaniline,5 poly(N,N′-di[p-phenylamino(phenyl)]-1,4,5,8-naphthalene tetracarboxylic diimide)6 and viologens7 have been widely used to construct electrochromic devices. Discrete transition metal complexes are a promising electrochromic alternative due to the vivid colours of their different oxidation states.8 Cobalt complexes of the cobaloxime family are under investigation as models for vitamin B129 and H2 evolution catalysts10 (Fig. 1a). We recently prepared compound [Co], which contains a cobaloxime core with two phosphonic acid anchor groups in the equatorial ligand framework for immobilisation on metal oxide surfaces (Fig. 1b). The good stability of [Co] on indium-tin oxide (ITO) and strong colouration for the [Co]I, [Co]II and [Co]III oxidation states11 encouraged us to investigate the potential use of [Co] in an electrochromic device.
![(a) Structure of [CoX(dimethylglyoximato)2(pyridine)] with X = CN− (Vit B12 model) and X = Cl− (H2 evolution catalyst). (b) Cobaloxime [Co] with phosphonic acid anchors for immobilisation.](/image/article/2013/CC/c3cc45881d/c3cc45881d-f1.gif) | ||
| Fig. 1 (a) Structure of [CoX(dimethylglyoximato)2(pyridine)] with X = CN− (Vit B12 model) and X = Cl− (H2 evolution catalyst). (b) Cobaloxime [Co] with phosphonic acid anchors for immobilisation. | ||
Recent developments in screen,12 microcontact13 and inkjet14 printing methodologies facilitate the necessary patterning of mesoporous electrode materials, such as semiconducting nanoparticle (NP) pastes. The former two techniques are however limited in the thicknesses of the transferred material, whereas inkjet printing is expensive, cumbersome and slow. Patterning of a NP film via a standard liftoff process has been demonstrated,15 but the resulting NP layer is thin (<1 μm).
In this communication, we present a lithographically structured tri-colour electrochromic device based on a facile liftoff process that is capable of patterning mesoporous ITO films with thicknesses up to 14 μm. Our approach is low-tech and does not require expensive equipment or materials. [Co] immobilised on these patterned ITO electrodes showed multi-coloured electrochromic performance.
First, a simple approach to patterned several micrometre thick mesoporous ITO electrodes was developed, which is schematically illustrated in Fig. 2 and described in detail in the ESI.† Fluoride-doped tin oxide (FTO) coated glass was cleaned in piranha solution (conc. H2SO4/30% H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3/1) at 90 °C for 10 min, followed by washing with water. An ITO NP (<40 nm) paste in acetic acid and EtOH was blade-coated onto the FTO. A negative photoresist (SU8) was sequentially spin-cast onto the ITO. The samples were soft-baked, exposed to UV light, post-baked and developed for 30 s with propylene glycol monomethyl ether acetate (soft- and post-bake procedure: 3 min at 65 °C, 3 min at 100 °C, and 3 min at 65 °C). The cross-linked SU8–ITO areas were lifted off by immersing in a mixture of tert-butanol and acetonitrile for 12 h (t-BuOH/ACN:1/1), followed by annealing in air at 450–550 °C for 20 min. This patterning approach was successful for film thicknesses ranging from 7 to 14 μm, with sub-millimetre features clearly visible on the patterned electrode (Fig. S1, ESI†).
:3/1) at 90 °C for 10 min, followed by washing with water. An ITO NP (<40 nm) paste in acetic acid and EtOH was blade-coated onto the FTO. A negative photoresist (SU8) was sequentially spin-cast onto the ITO. The samples were soft-baked, exposed to UV light, post-baked and developed for 30 s with propylene glycol monomethyl ether acetate (soft- and post-bake procedure: 3 min at 65 °C, 3 min at 100 °C, and 3 min at 65 °C). The cross-linked SU8–ITO areas were lifted off by immersing in a mixture of tert-butanol and acetonitrile for 12 h (t-BuOH/ACN:1/1), followed by annealing in air at 450–550 °C for 20 min. This patterning approach was successful for film thicknesses ranging from 7 to 14 μm, with sub-millimetre features clearly visible on the patterned electrode (Fig. S1, ESI†).
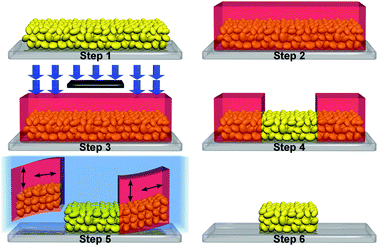 | ||
| Fig. 2 Schematic illustration of NP electrode patterning: (1) blade-coating of ITO onto piranha-cleaned FTO, (2) spin-casting of SU8, (3) UV-exposure, (4) development, (5) liftoff of the cross-linked SU8–ITO composite by soaking in t-BuOH/ACN, and (6) thermal annealing. | ||
The liftoff mechanism is based on the SU8–ITO bilayer architecture of the sacrificial areas. Swelling and deswelling of cross-linked SU8 upon t-BuOH/ACN solvent immersion and drying16 causes the delamination of the bilayer which is driven by tensile stresses induced by the SU8 top-layer, enabling a clean liftoff. The porous ITO areas are not subject to any deformation and stay unaffected during this process.
The mechanical stability of the sintered patterns was good, withstanding cleaning with a strong stream of nitrogen and prolonged immersion in organic solvents. [Co] was immobilised on the mesoporous ITO by soaking for 24 h in an anhydrous dimethylformamide (DMF) solution (6 mM of [Co]).
To investigate the electrochromic performance, transparent devices were assembled by capping the patterned ITO|[Co] with a FTO counter electrode using a precut thermoplastic gasket as the spacer (Fig. S2, ESI†). Propylene carbonate with 0.1 M tetrabutylammonium tetrafluoroborate (Bu4NBF4) or 1 M LiClO4 was used as electrolyte solvent rather than DMF which is classified as toxic and tends to soften the epoxy glue used for encapsulation. The three oxidation states of [Co] result in multi-coloured electrochromism (Fig. 3a and Fig. S3, ESI†). At an applied potential of −3.5 V the [Co]I exhibited a deep blue-green colour; at +3.0 V the yellow arises from [Co]III. Red colouration indicates the presence of an intermediate oxidation state [Co]II occurring at potentials of +1.5 V and −2.6 V during anodic and cathodic sweeps, respectively.
![(a) Photographs of an electrochromic display employing a 14 μm thick ITO|[Co] electrode. (b) Corresponding optical bright-field transmittance and dark-field reflectance spectra of the three oxidation states.](/image/article/2013/CC/c3cc45881d/c3cc45881d-f3.gif) | ||
| Fig. 3 (a) Photographs of an electrochromic display employing a 14 μm thick ITO|[Co] electrode. (b) Corresponding optical bright-field transmittance and dark-field reflectance spectra of the three oxidation states. | ||
The optical spectra of the three oxidation states for a 14 μm thick ITO|[Co] electrode are presented in Fig. 3b. The bright-field transmittance measurements were normalised with respect to the fully transparent area of the devices, whereas a white scattering surface below the FTO substrate was used as the reference for the dark-field reflectance measurements. The maximal optical contrasts at λ = 650 nm are ΔT = 28.3% and ΔR = 14.7% for transmittance and reflectance, respectively. Furthermore, these results were compared to the optical spectra of a 7 μm thick ITO|[Co] electrode (Fig. S4, ESI†). As expected, the thinner film exhibits a higher overall transmissivity compared to the 14 μm thick electrode, but the maximal optical transmission contrasts at λ = 650 nm are comparable. In contrast, an increased ITO|[Co] layer thickness dramatically enhances the colouration contrast in reflection.
Full colour electrochromic display applications require a gradually adjustable colouration control which can be achieved by applying a cell potential between −3.5 and +3.0 V (Fig. 4a). The [Co] oxidation state changes from III to I occur between −2.0 and −3.5 V during the cathodic cyclic voltammetry scan and are accompanied by a steep, S-shaped optical response at λ = 650 nm. The redox reactions and the corresponding colouration changes during the anodic sweep are more gradual. The transition from [Co]I to [Co]II occurs between −1.0 and +1.5 V whereas the further oxidation to [Co]III takes place at more positive potentials between +1.5 and +3.0 V. This in turn results in a step-wise colour change making the red [Co]II state more easily accessible. The cyclic voltammogram of [Co] in solution is presented in Fig. S5 (ESI†).
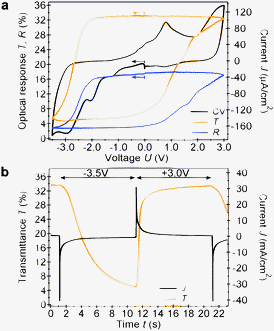 | ||
| Fig. 4 (a) Optical transmittance and reflectance response at λ = 650 nm of a 14 μm thick device and the corresponding cyclic voltammogram (10 mV s−1). Arrows indicate the scan direction and starting point (0 V). (b) Time-resolved optical response at λ = 650 nm and current density for an alternating step potential of −3.5 V and +3.0 V. | ||
A second important aspect of electrochromism is the temporal response upon alternating step potentials of −3.5 and +3.0 V (Fig. 4b). The ITO|[Co] electrode showed sharp and distinct transitions from the blue-green ([Co]I at −3.5 V) to the yellow ([Co]III at +3.0 V) state with a characteristic response time of 0.45 s. In contrast, the reverse step suffers from a slow, creeping optical response that only sets in after a dead time of 1 s followed by a characteristic response time of 2.7 s. Interestingly, the current curves of both these processes show the same initial peak that quickly decays. Furthermore, the cumulative charges passed through the cell during the 10 s potential steps are identical, exhibiting charge density values of ±17 mC cm−2. Assuming an electron conversion efficiency of 100%, this value relates to 8.8 × 10−8 mol of [Co] adsorbed per square-centimetre of a 14 μm thick electrode.
Cycle stability and stability of the liquid electrolyte encapsulation are further important requirements of electrochromic devices. Degradation of Bu4NBF4 quickly led to a brown precipitate, whereas the ITO|[Co] electrodes in combination with LiClO4 exhibited an improved long-term cycle stability over 500 switching cycles. On further cycling discolouration of the device due to degradation of the electrolyte became evident. A stand-by period of four months left the electrochromic performance unchanged and the ITO patterns as well as the cell seal intact.
In summary, we have reported the fabrication of a hybrid electrochromic device comprising the molecular cobaloxime complex [Co] immobilised on a patterned ITO electrode. The multi-coloured electrochromic performance arises from switching between the different oxidation states of the cobalt centre. Patterned mesoporous ITO films with thicknesses of up to 14 μm were fabricated using a modified lithography liftoff process, utilising low-tech fabrication steps and inexpensive equipment. On electrodes with individually addressable pixels, this can provide an electrochromic display that can access the RYB colour space.
References
- V. K. Thakur, G. Ding, J. Ma, P. S. Lee and X. Lu, Adv. Mater., 2012, 24, 4071–4096 CrossRef CAS PubMed.
- (a) M. R. J. Scherer and U. Steiner, Nano Lett., 2013, 13, 3005–3010 CrossRef CAS PubMed; (b) E. Unur, J.-H. Jung, R. J. Mortimer and J. R. Reynolds, Chem. Mater., 2008, 20, 2328–2334 CrossRef CAS; (c) A. L. Dyer, E. J. Thompson and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1787–1795 CrossRef CAS PubMed.
- (a) J. De Girolamo, P. Reiss, M. Zagorska, R. De Bettignies, S. Bailly, J.-Y. Mevellec, S. Lefrant, J.-P. Travers and A. Pron, Phys. Chem. Chem. Phys., 2008, 10, 4027–4035 RSC; (b) D. Aldakov, T. Jiu, M. Zagorska, R. de Bettignies, P.-H. Jouneau, A. Pron and F. Chandezon, Phys. Chem. Chem. Phys., 2010, 12, 7497–7505 RSC.
- K. Ding, H. Jia, S. Wei and Z. Guo, Ind. Eng. Chem. Res., 2011, 50, 7077–7082 CrossRef CAS.
- (a) C.-C. Wang, J.-F. Song, H.-M. Bao, Q.-D. Shen and C.-Z. Yang, Adv. Funct. Mater., 2008, 18, 1299–1306 CrossRef CAS; (b) H. Wei, X. Yan, S. Wu, Z. Luo, S. Wei and Z. Guo, J. Phys. Chem. C, 2012, 116, 25052–25064 CrossRef CAS.
- (a) H. Wei, X. Yan, Y. Li, H. Gu, S. Wu, K. Ding, S. Wei and Z. Guo, J. Phys. Chem. C, 2012, 116, 16286–16293 CrossRef CAS; (b) H. Wei, X. Yan, Y. Li, S. Wu, A. Wang, S. Wei and Z. Guo, J. Phys. Chem. C, 2012, 116, 4500–4510 CrossRef CAS; (c) Y. Li, R. Patil, S. Wei and Z. Guo, J. Phys. Chem. C, 2011, 115, 22863–22869 CrossRef CAS.
- N. Vlachopoulos, J. Nissfolk, M. Möller, A. Briançon, D. Corr, C. Grave, N. Leyland, R. Mesmer, F. Pichot, M. Ryan, G. Boschloo and A. Hagfeldt, Electrochim. Acta, 2008, 53, 4065–4071 CrossRef CAS PubMed.
- (a) P. Monk, R. Mortimer and D. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, 2007 Search PubMed; (b) J. A. MacCleverty, Comprehensive Coordination Chemistry II: From Biology to Nanotechnology, Elsevier, Pergamon, 2003 Search PubMed; (c) C. P. Horwitz and Q. Zuo, Inorg. Chem., 1992, 31, 1607–1613 CrossRef CAS; (d) H.-T. Zhang, P. Subramanian, O. Fussa-Rydel, J. C. Bebel and J. T. Hupp, Sol. Energy Mater. Sol. Cells, 1992, 25, 315–325 CrossRef CAS; (e) Q. Zeng, A. McNally, T. E. Keyes and R. J. Forster, Electrochem. Commun., 2008, 10, 466–470 CrossRef CAS PubMed; (f) D. J. Moore and T. F. Guarr, J. Electroanal. Chem. Interfacial Electrochem., 1991, 314, 313–321 CrossRef CAS; (g) H. Li and T. F. Guarr, J. Electroanal. Chem. Interfacial Electrochem., 1991, 297, 169–183 CrossRef CAS; (h) M. Kimura, T. Horai, K. Hanabusa and H. Shirai, Chem. Lett., 1997, 653–654 CrossRef CAS; (i) N. Trombach, O. Hild, D. Schlettwein and D. Wöhrle, J. Mater. Chem., 2002, 12, 879–885 RSC; (j) M.-a. Haga, T. Takasugi, A. Tomie, M. Ishizuya, T. Yamada, M. D. Hossain and M. Inoue, Dalton Trans., 2003, 2069–2079 RSC; (k) M. Biancardo, P. F. H. Schwab, R. Argazzi and C. A. Bignozzi, Inorg. Chem., 2003, 42, 3966–3968 CrossRef CAS PubMed; (l) M. Biancardo, R. Argazzi and C. A. Bignozzi, Displays, 2006, 27, 19–23 CrossRef CAS PubMed.
- G. N. Schrauzer, Acc. Chem. Res., 1968, 1, 97–103 CrossRef CAS.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2011, 136, 1067–1076 RSC.
- V. Santhanam, J. Liu, R. Agarwal and R. P. Andres, Langmuir, 2003, 19, 7881–7887 CrossRef CAS.
- (a) E. Tekin, P. J. Smith and U. S. Schubert, Soft Matter, 2008, 4, 703–713 RSC; (b) Q. Li and J. Lewis, Adv. Mater., 2003, 15, 1639–1643 CrossRef CAS.
- F. Hua, T. Cui and Y. Lvov, Langmuir, 2002, 18, 6712–6715 CrossRef CAS.
- (a) K. Wouters and R. Puers, J. Micromech. Microeng., 2010, 20, 095013 CrossRef; (b) E. J. Smith, D. Makarov and O. G. Schmidt, Soft Matter, 2011, 7, 11309–11313 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed description of the experimental techniques and additional transmission spectra. See DOI: 10.1039/c3cc45881d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2013 |