 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aromatic peptide amphiphiles: significance of the Fmoc moiety†
Scott Fleming*a, Sisir Debnatha, Pim W. J. M. Frederixab, Tell Tuttlea and Rein V. Ulijn*a
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK. E-mail: scott.fleming@strath.ac.uk; rein.ulijn@strath.ac.uk
bDepartment of Physics, University of Strathclyde, SUPA, Glasgow G4 0NG, UK
First published on 30th September 2013
Abstract
Aromatic peptide amphiphile hydrogelators commonly utilise the fluorenyl-9-methoxycarbonyl moiety as an N-terminal capping group. Material properties and spectroscopic techniques show the influence of alternative linkers between the fluorenyl moiety and the peptide. This study establishes whether methoxycarbonyl is an optimal or mainly convenient linker, for this class of self-assembling systems.
Aromatic peptide amphiphiles contain a short (e.g. di- or tri-) peptide sequence, with the N-terminus capped by a synthetic aromatic moiety.1 These compounds are commonly utilised in the preparation of supramolecular hydrogels. Here, self-assembly is based upon aromatic stacking interactions, and the propensity of the peptide to form a β-sheet type H-bonding arrangement.2–5 A variety of aromatic capping groups have been reported in the literature, such as naphthalene,6–8 azobenzene,9,10 and pyrene derivatives.11,12 However, by far the most commonly utilised functionality is the fluorenyl-9-methoxycarbonyl (Fmoc) group,13–19 due in part to its role as a protecting group in peptide synthesis.
A wide variety of Fmoc peptide amphiphiles are potent hydrogelators. However, the inherent cleavability of the carbamate linkage is not necessarily conducive to stable materials – particularly at high (e.g. 10.5) pH. Other peptide amphiphiles – such as those derived from naphthalene – seem to show a dependence upon the linker between the aromatic and peptide parts.7 In addition, we recently reported on the distinct infrared absorption spectra of Fmoc-dialanine and the related fluorenyl-9-methylcarbonyl-dialanine (Fmc-AA) hydrogels.20 Here, the Fmc-AA hydrogel was more difficult to prepare; being more sensitive to pH changes. Hence, we wanted to more generally ascertain whether or not Fmoc based hydrogelators could be readily replaced with closely related fluorene analogues without a significant impact upon the hydrogel's properties.
In this communication we study the self-assembly and hydrogelation characteristics of Fmoc-YL3,21 (3) and related fluorenyl compounds (Fig. 1). Fluorenyl-9-carbonyl-YL (1) has a minimal linker segment; fluorenyl-9-methylcarbonyl-YL (2) lacks the methoxy oxygen associated with 3; while in fluorenyl-9-ethylcarbonyl-YL (4) this oxygen is replaced with a methylene unit. These amide linkers were chosen as a means of investigating the relative impact of linker length and flexibility. DFT calculations on the geometry optimised structures 1, 2, 3, and 4 revealed molecular entropies (S) of 171.6, 178.0, 184.6, and 186.0 cal mol−1 K−1, respectively, demonstrating an increase of approximately 7–8 cal mol−1 K−1 per –CH2– (or –O–) group, mainly due to extra vibrational entropy (see ESI,† Table S1). It should be noted that there is a difference in the flexibility (entropy) associated with the carbamate moiety of 3, as the lone pairs on the methoxy oxygen, can potentially donate electron density to the adjacent carbonyl giving rise to a partial double bond (see ESI,† S5). Given the increased electronegativity of oxygen compared to nitrogen, this resonance form is not likely to be as significant as the analogous resonance structure that is well documented to restrict amide bond rotation.22,23 Nevertheless, our DFT calculations demonstrate an increased rotational energy barrier for 3 (12 kcal mol−1, see ESI,† S6) associated with the dihedral φ (indicated in Fig. 1) compared to that of 4 (3.7 kcal mol−1, see ESI,† S6). In addition, the methoxy oxygen is a potential H-bond acceptor, which could also have implications for the relative assembly characteristics of these materials.
 | ||
| Fig. 1 Fluorenyl based hydrogelator structures. | ||
All the compounds were found to form hydrogels at a 20 mM concentration in 100 mM sodium phosphate buffer. However, the required gelation pH differs considerably. 2 and 4 form hydrogels at the relatively acidic pH 6.5 and pH 6.0 respectively. Whereas in comparison, 1 and 3 successfully formed hydrogels at pH 7.3 – which was the highest hydrogelation pH attempted in this study. Successful gelation was initially determined by vial inversion, before the rheological properties were assessed via frequency sweep analysis (see ESI,† S1–S4). Of the physiological pH hydrogels, 3 was found to exhibit an elastic modulus of ∼390 Pa, whereas 1 was over an order of magnitude less rigid with an elastic modulus of ∼20 Pa. 2 and 4 exhibited elastic moduli of ∼230 Pa and ∼320 Pa respectively, and in addition exhibited a larger elastic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :viscous moduli ratio compared to gels 1 and 3. However, these properties are difficult to directly compare due to differences in the required gelation pH – at pH 7.3 systems 2 and 4 were solutions. These initial gelation results indicate that the choice of linker does have a significant impact upon the properties of these materials. Interestingly on the basis of these results 3 is the strongest gelator at physiological pH. The amide based linker analogues show a correlation between pH and linker length, with the shorter linkers being more optimal at high pH than the longer equivalents. These trends lend some credence to the notion that 3 behaves more like 1 because the carbamate is a relatively rigid linker on account of resonance arguments.
:viscous moduli ratio compared to gels 1 and 3. However, these properties are difficult to directly compare due to differences in the required gelation pH – at pH 7.3 systems 2 and 4 were solutions. These initial gelation results indicate that the choice of linker does have a significant impact upon the properties of these materials. Interestingly on the basis of these results 3 is the strongest gelator at physiological pH. The amide based linker analogues show a correlation between pH and linker length, with the shorter linkers being more optimal at high pH than the longer equivalents. These trends lend some credence to the notion that 3 behaves more like 1 because the carbamate is a relatively rigid linker on account of resonance arguments.
Fluorescence emission spectroscopy (Fig. 2) of the hydrogels shows that the most significant excimer redshift – indicative of extensive aromatic stacking interactions24,25 – is exhibited by 3, with an emission maximum around 450 nm. In comparison, 2 and 4, show multiple excimer bands around 380 and 400 nm – suggestive of less efficient aromatic overlap, previously attributed to antiparallel and parallel dimers.25 Interestingly, 1 does not show any excimer formation, instead only the redshift relative to the dilute solution spectrum (330 nm versus 315 nm) is observed. Hence, an extended J-aggregate is not inferred for this material. This is thought to be a consequence of the 1 linker being too short to allow orientation of the fluorenyl groups for effective aromatic stacking interactions. Generally, aromatic stacking is believed to be dominated by extended antiparallel fluorenyl–fluorenyl interactions, however, alternative less-aggregated fluorenyl arrangements and fluorenyl–tyrosine interactions are also likely to contribute to some extent. Overall, the amide linker trends are less clear, but evidently the Fmoc derivative 3 is found to exhibit the most extensive aromatic stacking arrangement.
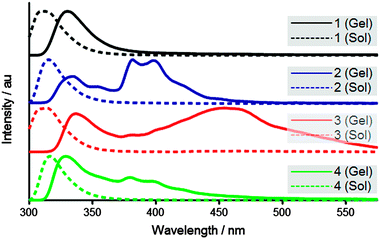 | ||
| Fig. 2 Fluorescence emission of gels (solid) and dilute solutions (dashed). | ||
Circular dichroism (Fig. 3) was used to probe the chiral orientation of the fluorenyl groups for each of the hydrogelator systems. All of the systems show a peak around 305 nm associated with the fluorenyl absorption.3,21 However, in terms of magnitude 3 and 4 exhibit a stronger molar ellipticity than the other systems. In addition, 3 also demonstrates substantial absorption bands at 275 nm, which may have some contribution from the tyrosine residue.26 In terms of the direction associated with the amide based linkers, it is of note that the handedness of the 305 nm peak switches between positive and negative as methylene units are added. This indicates that the length of the alkyl chain is having a direct effect upon the fluorenyl conformation, and the precise supramolecular chirality adopted. 3 appears to have a positive 305 nm peak similar to 2, which is interesting since both compounds possess a single methylene associated with their linker segments. These results again show the importance of the linker towards the self-assembly of these materials, and again suggest that 3 adopts a relatively ordered supramolecular arrangement.
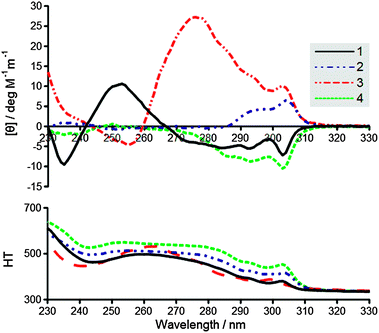 | ||
| Fig. 3 (top) Circular dichroism of gels, (bottom) high tension voltage. | ||
Infrared absorption spectroscopy (Fig. 4) of the gels (in D2O) in each case shows the amide I band around 1625 cm−1, associated with the increase in peak intensity due to the coupling of well-aligned H-bonded amide modes.20,27,28 However, the degree of apparent inhomogeneity associated with these materials varies considerably. While 1 and 3 exhibit relatively sharp well defined peaks, 2 and 4 demonstrate broader overlapping bands, suggestive of a variety of H-bonding stack sizes.18,29 These observations appear to qualitatively correlate with the required gelation pH in each case, suggesting more disorder associated with the lower pH hydrogels. In addition, it should be noted that 3 shows an additional band at 1685 cm−1 that is associated with the peak intensity enhancement of the aligned vibrational modes belonging to the carbamate moiety.20 As such this additional band is not believed to infer a distinct H-bonding arrangement associated with this material.
 | ||
| Fig. 4 Infrared absorption spectra of amide I region. Spectra are vertically offset for clarity. | ||
Atomic force microscopy (AFM) was used to examine the nanoscale morphology of the hydrogel networks (Fig. 5). As anticipated, fibres on the order of ∼40–200 nm in width were observed for 2, 3, and 4.3,30 However, significant differences between these systems were observed, with 2 showing very few fibres by AFM. In addition, no fibres were seen to be present in the corresponding sample of 1, which instead exhibited a more amorphous structure. These results should be interpreted with caution, as they are highly dependent upon the dried sample preparation, and as such aren't necessarily representative of the fibrous morphologies present in the gel state. Nevertheless, 3 appears to show the greatest propensity for fibre formation.
 | ||
| Fig. 5 Atomic force microscopy images of nanoscale morphology. | ||
In summary, we have shown that the linker between the fluorenyl moiety and the tyrosine–leucine (YL) dipeptide sequence has a significant impact upon the self-assembly and hydrogelation of these systems. The systems studied appear to suggest that hydrogelation is facilitated by more rigid linkers, with the Fmoc group falling into this category. Aromatic stacking interactions are strongly affected by the choice of linker, with short linkers restricting the conformation and aromatic stacking interactions associated with these materials. The handedness of the circular dichroism peaks also shows a clear dependence upon linker length. Overall, the Fmoc moiety appears to be the optimal choice for the YL dipeptide sequence – providing a rigid linker of sufficient length to allow effective assembly in both the aromatic and dipeptide domains. The implication being that Fmoc's role in peptide synthesis has been a fortuitous one for the field of small molecule self-assembly. In future, this work on linker optimisation will be extended to encompass a wider variety of aromatic groups, peptide sequences, and linkers possessing different heteroatoms in place of the methoxy oxygen.
Notes and references
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015–2022 CrossRef CAS PubMed.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- A. M. Smith, R. F. Collins, R. V. Ulijn and E. Blanch, J. Raman Spectrosc., 2009, 40, 1093–1095 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, J. Mater. Chem., 2007, 17, 850–854 RSC.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- Y. Lin, Y. Qiao, P. Tang, Z. Li and J. Huang, Soft Matter, 2011, 7, 2762–2769 RSC.
- Y. Huang, Z. Qiu, Y. Xu, J. Shi, H. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149–2155 CAS.
- Y. Zhang, Z. Yang, F. Yuan, H. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028–15029 CrossRef CAS PubMed.
- M. L. Ma, Y. Kuang, Y. Gao, Y. Zhang, P. Gao and B. Xu, J. Am. Chem. Soc., 2010, 132, 2719–2728 CrossRef CAS PubMed.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475–479 RSC.
- V. Castelletto, G. Cheng, B. W. Greenland, I. W. Hamley and P. J. F. Harris, Langmuir, 2011, 27, 2980–2988 CrossRef CAS PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–612 CrossRef CAS.
- G. Cheng, V. Castelletto, C. M. Moulton, G. E. Newby and I. W. Hamley, Langmuir, 2010, 26, 4990–4998 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–38 CrossRef CAS.
- S. Fleming, P. W. J. M. Frederix, I. Ramos-Sasselli, N. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- S. Roy, N. Javid, J. Sefcik, P. J. Halling and R. V. Ulijn, Langmuir, 2012, 28, 16664–16670 CrossRef CAS PubMed.
- W. D. Phillips, J. Chem. Phys., 1955, 23, 1363–1364 CrossRef CAS.
- A. H. Lewin and M. Frucht, Org. Magn. Reson., 1975, 7, 206–225 CrossRef CAS.
- H. K. Kang, D. E. Kang, B. H. Boo, S. J. Yoo, J. K. Lee and E. C. Lim, J. Phys. Chem. A, 2005, 109, 6799–6804 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- S. Bhattacharjee, G. Tóth, S. Lovas and J. D. Hirst, J. Phys. Chem. B, 2003, 107, 8682–8688 CrossRef CAS.
- H. Shao and J. R. Parquette, Chem. Commun., 2010, 46, 4285–4287 RSC.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- X. Mu, K. M. Eckes, M. M. Nguyen, L. J. Suggs and P. Ren, Biomacromolecules, 2012, 13, 3562–3571 CrossRef CAS PubMed.
- E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 8819–8820 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and analysis of compounds, methods, resonance structure diagram, and rheological frequency sweeps. See DOI: 10.1039/c3cc45822a |
| This journal is © The Royal Society of Chemistry 2013 |