 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly enantioselective hydrogenation and transfer hydrogenation of cycloalkyl and heterocyclic ketones catalysed by an iridium complex of a tridentate phosphine-diamine ligand†
José A. Fuentesa, Ian Carpentera, Nina Kannb and Matthew L. Clarke*a
aSchool of Chemistry, University of St Andrews, EaStCHEM, St Andrews, Fife, UK. E-mail: mc28@st-andrews.ac.uk; Fax: +(44) 1334 463808; Tel: +(44) 1334 463850
bDepartment of Chemical and Biological Engineering, Chalmers University of Technology, SE-412 96, Gothenburg, Sweden
First published on 23rd September 2013
Abstract
Ir complexes of chiral phosphine-diamine ligands catalyse the hydrogenation and transfer hydrogenation of aryl-piperidin-4-yl methanones, and ketones bearing both an aryl group and secondary alkyl substituent with up to 98% e.e., and with substrate to catalyst ratios of up to 4000.
The enantioselective reduction of ketones using molecular hydrogenation is one of the most important and scalable methods for preparing the ubiquitous chiral building blocks, secondary alcohols. The excellent results that are obtained in the reduction of a wide range of acetophenone derivatives using Noyori catalysts of type [RuCl2(diphos)(diamine)] are well known, and these Ru complexes are industrially viable catalysts (Fig. 1, 1).1 Most of these catalysts give between 85–99% e.e. and high rates for acetophenone derivatives. There has been considerable interest in examining both fundamentally different and slightly modified catalyst structures for ketone hydrogenation.2–5 This line of enquiry should, in principle lead to increased substrate scope. A case in point is the development of a range of Ru complexes of phosphine diamine and phosphine amino-alcohol ligands (Fig. 1, 2).3 These catalysts proved quite effective for the hydrogenation of α-tertiary alkyl ketones,3a heterocycle functionalised bulky ketones,3b,c esters3d,e and cyano-ketones.3f
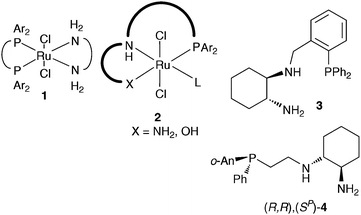 | ||
| Fig. 1 Schematic of the Noyori catalysts and the systems studied in St Andrews. | ||
Unfortunately, we have only been partially successful in rationally tuning the ligand structure to increase the ‘sweetspot’ in substrate scope for asymmetric hydrogenations.3g Using the Ru catalysts, we were unable to get synthetically useful results for some of our target applications, including the enantioselective hydrogenation of aryl(piperidin-4-yl)methanones. This was of interest to us, in part due to the steric bulk and free secondary amine presenting challenges, but more importantly due to their repeated occurrence as a chiral building block in the pharmaceutical patent literature.6 Motivated by the renaissance in the hydrogenation of carbonyl derivatives using iridium complexes,5 we have studied Ir catalysed ketone hydrogenation using phosphine-diamine ligands derived from cyclohexane-diamine. Here we show that changing from ruthenium to iridium, with the same very economic ligand system, delivers a catalyst that is able to hydrogenate this class of ketone to give the industrially important enantioenriched alcohols with high enantioselectivity. We also report an initial investigation into substrate scope that reveals this catalyst is highly competent for the reduction of a range of other substrates.
Due to the tendency of ligand 3 to give mixtures on reaction with well-known Ir precursors, and the observation that the best selectivity was observed with a ligand:iridium ratio of 0.5, an equimolar combination of [IrCl(COD)]2/(R,R)-3 was used as pre-catalyst in most of the reactions discussed below.
We examined the hydrogenation of the unprotected aryl(piperidin-4-yl)methanones 5k to 8k; these were readily prepared from the Weinreb amides of the piperidines.† While Rh catalysts have been used on chelating unprotected amino-ketones,4d hydrogenation of secondary amine-containing ketones generally requires protecting groups;1b,g,h the very few studies that specifically mention aryl(piperidin-4-yl)methanones use protected or tertiary amines.1h,7 We were therefore delighted to find that good conversions and high enantioselectivity can be obtained using this iridium catalyst. The dimethoxyphenyl substituted secondary alcohol has been the subject of large scale asymmetric synthesis studies before, from the Boc protected ketone 6k; 18 kilos of DIP-chloride chiral reducing agent were required to produce 6.45 Kg of product with 72.9% e.e. (4.6 Kg of 93% e.e. product after recryst.).6a Using either the Boc protected or the free NH compound and these new unoptimised catalytic protocols delivers product with higher selectivity with significantly better atom economy. The Boc protected ketone seems to reduce somewhat faster, but with lower selectivity in the hydrogenations (Table 1, entries 2 and 4). Aryl(piperidin-4-yl)methanones with less bulky aryl groups gave yet higher selectivity (Table 1, entries 6–10), reaching close to enantiopurity (Scheme 1).
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | T (°C) | Time (h) | Conversion (%) [yield] | ee |
| a Reactions carried out using 0.1 mol% [IrCl(COD)]2 and 0.1% (R,R)-3 using 2 mol% KOtBu co-catalyst in dry IPA at 50 bar initial hydrogen pressure, unless indicated otherwise. See ESI for determination of ee and configurations.b (R,R)-3 gives S product.c 0.05 mol% [IrCl(COD)]2 0.1% 3. | |||||
| 1 | 5k | 35 | 71 | 70 | 80b |
| 2 | 5k | 50 | 25 | >99 [68] | 78 |
| 3 | 6k | 17 | 48 | >99 [94] | 75 |
| 4 | 6k | 50 | 17 | >99 | 75 |
| 5 | 6k | 50 | 17 | >99 | 69c |
| 6 | 7k | 35 | 71 | >99 [71] | 94 |
| 7 | 7k | 15 | 65 | 75 | 93 |
| 8 | 8k | 22 | 42 | >99 [77] | 96 |
| 9 | 8k | 50 | 14 | >99 [72] | 93 |
| 10 | 8k | 50 | 2 | >99 | 96 |

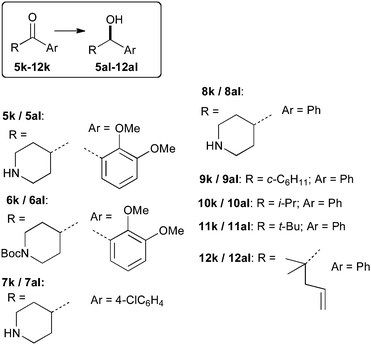 | ||
| Scheme 1 Substrates (k) reduced to alcohols (al) in this study. | ||
In order to investigate if this high selectivity was a very specific niche for piperidine functionalised ketones, a range of studies were carried out on ketones 9k–12k. The results in Table 2 show that using 0.1% of the new Ir system, the cyclohexyl substituted ketone, 9k can be reduced very selectively: 98% e.e. and high conversion are obtained in 20 minutes at room temperature (Table 2, entry 5). While we have not carried out a full scale up study, we note that, even using a magnetically stirred reaction vessel, high conversion for the hydrogenation of 9k can be reached using 0.025 mol% catalyst loading with 95% e.e.; a single unoptimised recrystallisation gave enantiopure material in 61% yield. Isobutyrophenone, 10k was also reduced with high enantioselectivity (Table 2, entry 10). The tertiary alkyl substituted ketones gave significantly inferior results than those obtained with Ru catalysts of ligand 3. However, rather more encouraging results could be obtained when ligand 3 is substituted with P-chiral ligand 4. For example, the successful and completely chemoselective hydrogenation of the unsaturated ketone 12k at 0.1 mol% catalyst loading with significantly better e.e. than with the parent system (Table 2: compare entries 13 and 14) shows that this type of catalyst can be tuned and therefore bodes well for other applications.
| Entry | Substrate | Catalyst (%) ligand [%] | T (°C) | Time (h) | Conversion (%) [yield] | ee |
|---|---|---|---|---|---|---|
| a Reactions carried out using 0.1 mol% [IrCl(COD)]2 and 0.1% ligand (R,R)-3 using 2 mol% KOtBu co-catalyst in dry IPA at 50 bar initial hydrogen pressure, unless indicated otherwise. See ESI for determination of ee and configurations.b 0.025% [IrCl(COD)]2, 0.25% KOtBu, 61% yield, >99% e.e.c Complex 13 was used.d Ligand 4 was used. | ||||||
| 1 | 9k | 0.05 [0.1] | 50 | 17 | >99 | 77(S) |
| 2 | 9k | 0.1 [0.1] | 50 | 17 | >99 [89] | 84(S) |
| 3 | 9k | 0.05 [0.05] | 80 | 2 | 89 | 90(S) |
| 4b | 9k | 0.025 [0.025] | 60 | 46 | 89 | 95(S) |
| 5 | 9k | 0.1 [0.1] | 21 | 0.33 | 78 | 98(S) |
| 6c | 9k | 0.1 | 21 | 0.33 | 85 | 97(S) |
| 7 | 9k | 0.1 [0.1] | 45 | 1 | 98 | 93(S) |
| 8c | 9k | 0.1 | 45 | 1 | 97 | 87(S) |
| 9c | 9k | 0.1 | 50 | 3 | 98.5 | 64(S) |
| 10 | 10k | 0.5 [0.5] | 50 | 1 | >99 [97] | 89(S) |
| 11 | 11k | 0.05 [0.1] | 50 | 22 | 99 | 18.5(S) |
| 12 | 11kd | 0.05 [0.1] | 50 | 22 | 98 [89] | 67(S) |
| 13 | 12k | 0.1 [0.1] | 17 | 48 | 96 | 32 |
| 14 | 12kd | 0.05 [0.1] | 40 | 22 | 99 [80] | 71 |
All our attempts at reacting ligand 3 with [IrCl(COD)]2 or [Ir(COD)2]+ at various stoichiometries gave mixtures that could not be characterised and also decomposed within hours, even in degassed solvents. Many attempts at growing crystals suitable for X-ray diffraction failed. A complex that was free of any obvious impurities could be isolated from the reaction of [Ir(OMe)(COD)]2 and ligand 3. The spectroscopic data is quite strongly supportive of an amido complex with a formula [Ir(L–H)(COD)] (13) in which ligand 3 exhibits a tridentate anionic coordination mode. Further discussion of this assignment is in the ESI.† With this complex and the in situ system in hand, we have carried our preliminary studies to understand the nature of this reduction, which is carried out in IPA solvent. Complex 13 and [IrCl(COD)]2/3 also catalyse transfer hydrogenation using IPA as hydrogen source. These reactions give similarly high selectivity and productivity as the hydrogenations, and isolated complex 13 behaves similarly to the in situ catalyst (Scheme 2 and further examples in ESI,† Table S3).
 | ||
| Scheme 2 Transfer hydrogenation using the Ir–phosphinediamine system is rapid and highly selective. | ||
The hydrogenation reactions do utilise molecular hydrogen as reductant as confirmed by an experiment using a ballast vessel at constant pressure, and enhanced conversion at room temperature relative to transfer hydrogenation.† The stoichiometric reaction of 13 with hydrogen in the absence of IPA gives cyclooctane (from hydrogenation of COD), and several Ir-hydride species as determined by the multitude of peaks in the hydride region of the 1H NMR. The amido complex also reduces ketones very slowly but selectively without added base.† The gas uptake of two complex-13-catalysed reductions of 9k at 24 and 10 bar show that the reactions are zero order in substrate and positive order in hydrogen.† This was confirmed for both 13 and the in situ catalyst with separate experiments run at 0.25 and 0.5 M concentration at 50 bar pressure. The independence on substrate concentration, implying that hydride transfer is faster than other steps in the cycle (i.e. activation of hydrogen), is in contrast to the slower reactions catalysed by Ru complexes of P, NH, NH2 ligands.3c This shows that the Ir systems are more proficient at the transfer of hydride to the ketone. Complex 13 gives significantly lower selectivity than [IrCl(COD)]2/3 under pressure hydrogenation reactions at higher temperatures (e.g. 64% e.e. 99% conv. after 3 hours at 50 °C, 0.1% 13), but gives similar results at room temperature (e.g. 96% e.e. at 81% conv. after 20 minutes at 21 °C, 0.1% 13). For the [IrCl(COD)]2/3 system the e.e. does not degrade too much from 98% maximum right up to 80 °C (Table 2). A full table (20 expts) is provided in the ESI.† A hydrogenation carried out in d8-IPA reveals that over 90% of the CD(OH/D) signal is deuterated and hence the transfer hydrogenation is competitive with pressure hydrogenation. While since it is possible that Ir catalysed H–D exchange can occur, but this is only possible when the fundamental steps of transfer hydrogenation are competitive with those of pressure hydrogenation. In the Ru catalysed reductions, less than 20% of deuterium incorporation is observed3a in accordance with those catalysts being much slower at transfer hydrogenation.
In conclusion, the combination of [IrCl(COD)]2 and chiral P, N, N ligands derived from cyclohexane diamine gives a catalyst that exhibits high selectivity and activity in the hydrogenation and transfer hydrogenation of ketones bearing an aryl substituent and some form of secondary alkyl group. The selectivity and activity observed is significantly higher than that obtained with a Ru catalyst derived from the same ligand (or in fact the use of [RhCl(COD)]2/ligand 3.†). The ability to deliver >90% e.e. at low catalyst loading and near ambient temperature for these more challenging substrates means this catalyst is already of synthetic use, but this type of iridium catalyst is also a worthwhile lead structure for further research.
The authors thank the EPSRC National Mass Spectrometry Service, the St Andrews technical staff for their assistance, the EPSRC, the Swedish Research Council, and the University of St Andrews, for funding. MLC also thanks the Royal Society for an Industry Fellowship and Dr Reddys Laboratories (UK) for their co-support of this.
Notes and references
- (a) T. Ohkuma, S. Hashaguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675 CrossRef CAS; (b) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS; (c) p. Saji, in The handbook of homogeneous hydrogenation, ed. C. J. Elsevier and J. G. De Vries, Wiley-VCH, Weinheim, 2007 Search PubMed; (d) T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori and K. Murata, Org. Lett., 2007, 9, 255 CrossRef CAS PubMed; (e) T. Ohkuma, C. A. Sandoval, R. Srinivasan, Q. Lin, Y. Wei, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2005, 127, 8288 CrossRef CAS PubMed; (f) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724 CrossRef CAS PubMed; (g) M. Akashi, N. Arai, T. Inoue and T. Ohkuma, Adv. Synth. Catal., 2011, 353, 1955 CrossRef CAS; (h) N. Arai, H. Ooka, K. Azuma, T. Yabuuchi, N. Kurono, T. Inoue and T. Ohkuma, Org. Lett., 2007, 9, 939 CrossRef CAS PubMed; (i) N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291 CrossRef CAS PubMed.
- Some chiral catalysts with different designs: (a) Y. J. Xu, N. W. Alcock, G. J. Clarkson, G. Docherty, G. Woodward and M. Wills, Org. Lett., 2004, 6, 4105 CrossRef CAS PubMed; (b) Y. Xu, G. C. Clarkson, G. Docherty, C. L. North, G. Woodward and M. Wills, J. Org. Chem., 2005, 70, 8079 CrossRef CAS PubMed; (c) Q. Jing, X. Zhang, H. Sun and K. L. Ding, Adv. Synth. Catal., 2005, 347, 1193 CrossRef CAS; (d) F. Naud, C. Malan, F. Spindler, C. Ruggeberg, A. T. Schmidt and H. U. Blaser, Adv. Synth. Catal., 2006, 348, 47 CrossRef CAS; (e) K. Abdur-Rashid, R. W. Guo, A. J. Lough, R. H. Morris and D. T. Song, Adv. Synth. Catal., 2005, 347, 571 CrossRef CAS; (f) H. Huang, T. Okuno, K. Tsuda, M. Yoshimura and M. Kitamura, J. Am. Chem. Soc., 2006, 128, 8716 CrossRef CAS PubMed; (g) N. Arai, K. Suzuki, S. Sugizaki, H. Sorimachi and T. Ohkuma, Angew. Chem., Int. Ed., 2008, 47, 1770 CrossRef CAS PubMed; (h) W. N. M. O, A. J. W. Lough and R. H. Morris, Chem. Commun., 2010, 46, 8240 RSC.
- (a) M. L. Clarke, M. B. Diaz-Valenzuela and A. M. Z. Slawin, Organometallics, 2007, 26, 16 CrossRef CAS; (b) M. B. Diaz-Valenzuela, S. D. Phillips, M. B. France, M. E. Gunn and M. L. Clarke, Chem.–Eur. J., 2009, 15, 1227 CrossRef CAS PubMed; (c) S. D. Phillips, J. A. Fuentes and M. L. Clarke, Chem.–Eur. J., 2010, 16, 8002 CrossRef CAS PubMed; (d) I. Carpenter, S. C. Eckelmann, M. T. Kuntz, J. A. Fuentes, M. B. France and M. L. Clarke, Dalton Trans., 2012, 41, 10136 RSC; (e) M. L. Clarke, Catal. Sci. Technol., 2012, 2, 2418 RSC; (f) J. A. Fuentes, S. D. Phillips and M. L. Clarke, Chem. Cent. J., 2012, 6, 151 CrossRef CAS PubMed; (g) S. D. Phillips, K. H. O. Anderson, N. Kann, M. T. Kuntz, M. B. France, P. Wawryzniak and M. L. Clarke, Catal. Sci. Technol., 2011, 1, 1336 RSC.
- Other metals used in asymmetric ketone hydrogenation include: (a) Os catalysts: W. Barratta, M. Ballico, G. Chelucci, K. Siega and P. Rigo, Angew. Chem., Int. Ed., 2008, 47, 4362 CrossRef PubMed; (b) Fe catalysts: R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (c) Cu catalysts: K. Junge, B. Wendt, D. Addis, S. L. Zhou, S. Das, S. Fleischer and M. Beller, Chem.–Eur. J., 2011, 17, 101 CrossRef CAS PubMed and ref's therein; ; (d) Rh catalysis: T. Hayashi and M. Kumada, Acc. Chem. Res., 1982, 15, 395 CrossRef CAS.
- (a) J.-B. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538 CrossRef CAS PubMed; (b) J. E. D. Martins, D. J. Morris and M. Wills, Tetrahedron Lett., 2009, 50, 688 CrossRef CAS; (c) T. Irrgang, D. Friedrich and R. Kempe, Angew. Chem., Int. Ed., 2011, 50, 2183 CrossRef CAS PubMed; (d) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967 CrossRef CAS PubMed; (e) transfer hydrogenation using Ir is more common, see R. J. Lundgren and M. Stradiotto, Chem.–Eur. J., 2008, 14, 10388 CrossRef CAS PubMed and ref's therein; (f) R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729 CrossRef CAS; (g) J. K. Liu, X. F. Wu, J. A. Iggo and J. L. Xiao, Coord. Chem. Rev., 2008, 252, 782 CrossRef CAS; (h) W. W. N. O, A. J. Lough and R. H. Morris, Organometallics, 2012, 31, 2152 CrossRef CAS; (i) J.-B. Xie, J.-H. Xie, X.-Y. Liu, Q. Q. Zhang and Q.-L. Zhou, Chem.–Asian J., 2011, 6, 899 CrossRef CAS PubMed.
- (a) E. D. Daugs, J. C. Evans, H.-W. Freming, T. H. E. Hilpert, J. N. Koek, F. M. Laskovics, S. K. Stolz-Dunn and I. A. Tomlinson, US20020151717, Aventis, 2002; (b) S. K. Stolz-Dunn, J. Evans and I. A. Tomlinson, US7332607, Aventis, 2008; (c) R. B. Diebold, G. Thomas, P. Grover, S. Huang, S. Ionndis, C. A. Ogoe, J. C. Saeh and J. Varnes, WO2012/017252A1, Astra Zeneca, 2011; (d) G. J. Bridger, Y. Zhou and R. Skerlj, WO2005/059107, Anormed Inc., 2004; (e) F. Ledo-Gomez, A. Munoz Munoz and C. Pumar Duran, WO2011/147780A1, Faes Farma S. A., 2011.
- D. J. Morris, A. M. Hayes and M. Wills, J. Org. Chem., 2006, 71, 7035 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, analytical data, NMR and HPLC spectra. See DOI: 10.1039/c3cc45545a |
| This journal is © The Royal Society of Chemistry 2013 |