 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Clickable” LNA/DNA probes for fluorescence sensing of nucleic acids and autoimmune antibodies†
Anna S. Jørgensen, Pankaj Gupta, Jesper Wengel and I. Kira Astakhova*
Nucleic Acid Center, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, DK-5230 Odense M, Denmark. E-mail: ias@sdu.dk; Fax: +45 6615 8780; Tel: +45 6550 2510
First published on 27th September 2013
Abstract
Herein we describe fluorescent oligonucleotides prepared by click chemistry between novel alkyne-modified locked nucleic acid (LNA) strands and a series of fluorescent azides for homogeneous (all-in-solution) detection of nucleic acids and autoimmune antibodies.
Several autoimmune disorders are characterized by production of antibodies against single- and double-stranded DNA. If not diagnosed and treated early, the autoimmune conditions can lead to serious health deterioration and even mortality.1 The sequence-specific autoimmune antibodies (autoantibodies) against single-stranded DNA have been thoroughly studied.2 In turn, non-sequence-specific autoantibodies against double-stranded DNA, a hallmark of autoimmune conditions such as antiphospholipid syndrome and systemic lupus erythematosus (SLE), have not been studied in detail.1
Generally, monitoring interactions of nucleic acids by fluorescence is a convenient method in modern bioanalysis and can be performed under native conditions without additional equipment or procedures. Currently, fluorescent oligonucleotides containing bright cyanine and xanthene dyes are often applied in bioanalysis of nucleic acids3 and proteins, including antibodies.4 Furthermore, affinity-enhancing locked nucleic acids containing 2′-amino-LNA monomers with fluorescent polyaromatic hydrocarbons (PAHs) attached at the 2′-amino group provide high target binding affinity and selectivity, remarkable fluorescence quantum yields and brightness values.5 Another appealing aspect of LNA/DNA probes is their high potential as aptamers in selective binding of diverse proteins.6
Herein, a new alkyne-LNA nucleotide M1 incorporated into synthetic oligonucleotides was used in copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC or “click”)7 reactions with xanthene, cyanine and PAH azides 5–8 (Scheme 1). We describe preparation and the bioanalytical potential of the resulting LNA/DNA probes in fluorescence homogeneous sensing of nucleic acids and autoantibodies against double-stranded DNA, as well as structural aspects of the interactions clarified by the developed fluorescence assay.
 | ||
| Scheme 1 Synthesis of alkyne-LNA monomer M1 and incorporation of monomers M1–M5 into synthetic oligonucleotides. | ||
Previously, satisfactory biosensing properties of fluorescent probes labelled at the 2′-position of uridine by CuAAC reactions were demonstrated.8 In the present study we designed alkyne-LNA monomer M1 which combines the unique bicyclic structure of 2′-amino-LNA5 with a terminal alkyne group, allowing post-synthetic attachment of different tags by click chemistry.
Monomer M1 was incorporated into oligonucleotides using the phosphoramidite building block 3 which was prepared starting from the corresponding 2′-amino-5′-O-dimethoxytrityl protected LNA nucleoside 1 in two steps with 52% overall yield. Subsequent automated DNA synthesis furnished modified 21-mer oligonucleotides similar to those previously used in studies of 2′-alkyne-uridine (Scheme 1 and Table 1).10 After purification and characterization by ion-exchange (IE) HPLC and MALDI-MS (ESI†), ON1–ON4 were subjected to CuAAC reactions with fluorescent azides of three important classes: xanthene 5-R110 (5), cyanines Cy3 and Cy5 (6 and 7, respectively), and PAH perylene 8 (Table S1, ESI†). Conditions of the click chemistry were adjusted for each azide, taking into account the high hydrophobicity of the cyanine and perylene dyes, giving 62–81% yields of the products in ≥95% purity as determined by IE HPLC (Table S2, ESI†).
| ON | Sequence, 5′ → 3′ | Tm/ΔTm (°C) | |
|---|---|---|---|
| Target | |||
| DNA | RNA | ||
| a Thermal denaturation temperatures Tm (°C)/change in Tm relative to the corresponding unmodified duplex, ΔTm (°C) (ESI). | |||
| DNAref | TGCACTCTATGTCTGTATCAT | 59.0 | 60.5 |
| ON1 | TGCACTCTATG![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif) 1CTGTATCAT 1CTGTATCAT | 62.0/+3.0 | 65.0/+4.5 |
| ON2 | TGCACT CTAM1GTC1GTAT CAT | 63.5/+4.5 | 69.0/+8.5 |
| ON3 | TGCAC1CTATGTCTGTA1CAT | 63.0/+4.0 | 68.0/+7.5 |
| ON4 | TGCAC1CTATG1CTGTA1CAT | 65.0/+6.0 | 71.0/+10.5 |
| ON5 | TGCACTCTATG2CTGTATCAT | 59.0/0.0 | 62.0/+1.5 |
| ON6 | TGCACT CTA2GTC2GTAT CAT | 60.0/+1.0 | 66.0/+5.5 |
| ON7 | TGCAC2CTATGTCTGTA2CAT | 58.0/−1.0 | 64.0/+3.5 |
| ON8 | TGCAC2CTATG2CTGTA2CAT | 58.0/−1.0 | 66.0/+5.5 |
| ON9 | TGCACTCTATG3CTGTATCAT | 62.0/+3.0 | 63.0/+2.5 |
| ON10 | TGCACT CTA3GTC3GTAT CAT | 63.0/+4.0 | 66.0/+5.5 |
| ON11 | TGCAC3CTATGTCTGTA3CAT | 63.0/+4.0 | 65.0/+4.5 |
| ON12 | TGCAC3CTATG3CTGTA3CAT | 65.0/+6.0 | 66.5/+6.0 |
| ON13 | TGCACTCTATG4CTGTATCAT | 62.0/+3.0 | 63.0/+2.5 |
| ON14 | TGCACT CTA4GTC4GTAT CAT | 64.0/+5.0 | 65.0/+4.5 |
| ON15 | TGCAC4CTATGTCTGTA4CAT | 63.0/+4.0 | 64.5/+3.5 |
| ON16 | TGCAC4CTATG4CTGTA4CAT | 65.0/+6.0 | 65.5/+5.0 |
| ON17 | TGCACTCTATG5CTGTATCAT | 66.0/+7.0 | 65.0/+4.5 |
| ON18 | TGCACT CTA5GTC5GTAT CAT | 70.0/+11.0 | 60.0/−0.5 |
| ON19 | TGCAC5CTATGTCTGTA5CAT | 67.0/+8.0 | 67.0/+6.5 |
| ON20 | TGCAC5CTATG5CTGTA5CAT | 72.0/+13.0 | 70.0/+9.5 |
Binding affinity of ON5–ON20 to complementary and mismatched DNA/RNA targets was evaluated in a medium salt phosphate buffer ([Na+] = 110 mM, pH 7.0) by thermal denaturation (Tm) measurements monitoring absorbance at 260 nm and at the characteristic fluorophores' wavelength (Tables S3–S5, ESI†). First, the resulting Tm values were similar at both wavelengths suggesting high sensitivity of the dyes to hybridization. Second, affinity enhancing LNA monomers resulted in high Tm values of the duplexes with complementary DNA/RNA suggesting that the fluorophores are tolerated within the double strands as described earlier for other fluorophores.8 Further, especially high target binding was displayed by the probes having M5 (ΔTm up to +13 °C for three incorporations), most likely provided by additional interactions involving the attached perylene groups. Third, thermal denaturation values for the selected probes ON7 and ON19 were decreased by 8–18 °C in the presence of a single mismatch in target DNA/RNA, confirming high binding selectivity for the examined probes (Table S4, ESI;† the probes were selected based on their attractive fluorescence properties as potential biosensors described below).
Notably, hybridization resulted in hypsochromic shift of absorbance maxima by the monomer M2 (Δλabsmax 7–9 nm), confirming placement of the xanthene within the minor groove of the duplexes.8 In turn, M3–M5 showed minor changes in absorbance peaks upon binding DNA/RNA (Δλabsmax 1–3 nm), although in the case of the cyanine monomers M3–M4 the ratio between two visible absorbance bands (RI\II) increased upon hybridization. According to the literature9 this indicates reduced dye interactions upon hybridization and their aggregation within single-stranded probes (Fig. S5, ESI;†e.g. RI\II 1.0 and 2.3 for ON16 and ON16:RNA, respectively).
Steady-state fluorescence emission spectra of ON5–ON20 and their duplexes with DNA/RNA targets were recorded in a medium salt buffer at 19 °C. In order to evaluate the effect of internal attachment of the fluorophores on fluorescence properties of the probes, their fluorescence intensities were compared to those of 5′-labelled LNA/DNA references (Fig. 1; Table S1, ESI†). As confirmed by fluorescence spectra, large hydrophobic dyes M2–M4 aggregated in a highly polar environment of the single-stranded probes, which resulted in quenched fluorescence (Table S6, ESI;† fluorescence quantum yields ΦF 0.01–0.16).8,9 Similarly, fluorescence of the duplexes possessing internally incorporated cyanine monomers M3 and M4 was low (ΦF 0.01–0.11), most likely due to active interactions of the dyes and nucleic acids.8 In contrast, fluorescence intensity of single-stranded probes and duplexes containing monomer M5 was extraordinarily high (ΦF 0.54–1.00 and fluorescence brightness (FB) values up to 80). Efficient sensing of hybridization accompanied by high ΦF and FB values was displayed exclusively by the probes ON7 and ON8 having double and triple incorporation of the monomer M2, respectively (Fig. 2; ESI†). Upon binding complementary targets by ON7 and ON8, up to 7.8-fold light-up of fluorescence was observed at λflmax 530 nm, accompanied by ΦF 0.22–0.45 and FB values up to 77 of the corresponding duplexes. Being compared to commercially available 5′-modified LNA/DNA references (Fig. 2) and other fluorescent probes,3,8,9 the probes prepared herein generally display improved binding affinity, sensing of hybridization, fluorescence quantum yield and brightness values, which are important properties for their application in various bioanalytical assays. Finally, fluorescence intensities of duplexes formed by ON7 and ON19 decreased 1.9–9.9 times in the presence of single-base mismatches in target DNA/RNA (Table S4, ESI†). Decreased emission in the presence of a mismatch is most likely caused by high sensitivity of the fluorescence of the internally incorporated monomers M2 and M5 to the local microenvironment within the biomolecules.8
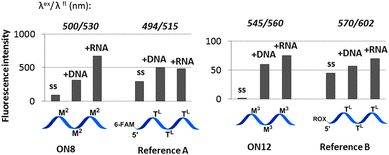 | ||
Fig. 1 Representative fluorescence intensities of the probes (ss) and duplexes compared to 5′-labelled LNA/DNA references. Spectra were obtained in a medium salt buffer at 19 °C using 1.0 μM oligonucleotides. Reference A: 5′-(6-FAM)-d(TGC AC![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) L CTA TG L CTG TAL CAT)-3′; reference B: 5′-ROX-d(TGC ACL CTA TG L CTG TAL CAT)-3′. TL = LNA-T nucleotide. L CTA TG L CTG TAL CAT)-3′; reference B: 5′-ROX-d(TGC ACL CTA TG L CTG TAL CAT)-3′. TL = LNA-T nucleotide. | ||
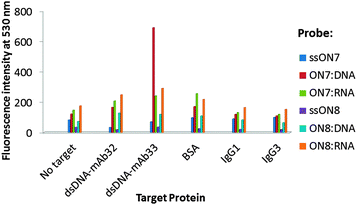 | ||
| Fig. 2 Fluorescence detection of monoclonal autoantibodies (ESI†). | ||
An important advantage of synthetic oligonucleotides within molecular diagnostics of proteins (so-called aptasensing approach) and immunoimaging techniques is their high specificity in binding a target.10 To assess the potential of the novel probes in diagnostics of clinically important proteins, fluorescence homogenous detection of human autoantibodies against double-stranded DNA was performed. Single-stranded ON7–ON8 and their duplexes with complementary DNA/RNA were incubated with commercially available human monoclonal autoantibodies dsDNA-mAb32 and dsDNA-mAb33, which were recently studied by surface plasmon resonance (SPR).10 The subtypes of the monoclonal antibodies were IgG1 (dsDNA-mAb33) and IgG3 (dsDNA-mAb32), and both antibodies have been used as serological parameters in diagnostics of SLE.11
Incubation was then performed in a medium salt phosphate buffer at 37 °C for 1 h followed by analysis after 2 h at 19 °C (ESI†). In order to evaluate the probes' specificity bovine serum albumin (BSA) and non-specific isotype antibodies IgG1 and IgG3 were used as references.12 Unlike single-stranded ON7 and other examined complexes, ON7:DNA showed 5.7-fold increase of fluorescence at 530 nm when binding dsDNA-mAb33, and 4.2-fold greater fluorescence than in the presence of dsDNA-mAb32, BSA or IgG controls (Fig. 2). Previously SPR studies indicated weaker binding for dsDNA-mAb33 compared to dsDNA-mAb32 by a 24 bp DNA duplex ((kd)obs ∼ 6.5 × 10−3 s−1 and 0.5 × 10−3 s−1, respectively).10 Thus, the binding pattern of ON7:DNA implies that chemical modification might change binding properties of nucleic acids to target proteins. In contrast to ON7:RNA and triply modified ON8:DNA/RNA, little to no fluorescence signal of interaction with BSA or non-specific isotype IgGs was observed for ON7:DNA, confirming high binding selectivity for the latter complex (Table S7, ESI†). According to our molecular models, triple incorporation of the xanthene dyes (M2) results in high surface hydrophobicity of the duplexes, which may account for their non-specific binding to BSA (ESI;† Fig. S7a). In turn, effective recognition of dsDNA-mAb33 is provided by steric and chemical complementarity of the unmodified internal region of ON7:DNA and the variable region of the autoantibody's heavy chain, accompanied by effective hydrogen bonding (Fig. S7b, ESI,† compared to c and d). We speculate that similarly to hybridization described above, target binding results in positioning of the xanthenes in a less polar environment compared to the initial nucleic acid complex resulting in an increased fluorescence.
Finally, the limit of target detection (LOD) for ON7:DNA was determined to be below 4.6 μg mL−1 of dsDNA-mAb33 (Fig. S8, ESI†). This is comparable with currently applied enzyme-linked immunosorbent assay (ELISA), immunofluorescence tests (LOD approx. 1–2 μg mL−1),1 and other fluorescent aptasensors.13 Notably, being compared to voltage current and electrochemical methods, homogeneous detection is robust, rapid and does not affect interacting surfaces of the biomolecules which can be detected without the need for additional steps and reagents.13,14
In conclusion, the click approach presented here efficiently yields probes with various dyes attached internally at the 2′-amino-position of 2′-amino-LNA monomers. This approach provides a reliable foundation for simple and efficient preparation of a library of fluorescent probes, screening and identification of several bright oligonucleotides with high target binding affinity and specificity. As demonstrated by our initial studies, potential applications of these probes include a wide range of fluorescence assays including, but not limited to, live-cell nucleic acid imaging, aptasensing and nucleic acid diagnostics. Moreover, the 2′-N-alkynyl group in the LNA/DNA strands is available for the attachment of other tags such as carbohydrates, lipids, cofactors and cell-penetrating peptides. In this context we believe that “clickable” LNA/DNA probes offer appealing opportunities for developing efficient tools for biosensing, pharmacological or nano-production purposes.
The authors would like to acknowledge financial support from The Sapere Aude programme of The Danish Council for Independent Research, THE VILLUM FOUNDATION and The European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC Grant agreement No. 268776.
Notes and references
- B. Giannakopoulos, F. Passam, Y. Ioannou and S. A. Krilis, Blood, 2009, 113, 985 CrossRef CAS PubMed.
- P. C. Ackroyd, J. Cleary and G. D. Glick, Biochemistry, 2001, 40, 2911 CrossRef CAS PubMed.
- M. D. Blower, E. Feric, K. Weis and R. Heald, J. Cell Biol., 2007, 179, 1365 CrossRef CAS PubMed.
- S. C. B. Gopinath, K. Awazu and M. Fujimaki, Sensors, 2012, 12, 2136 CrossRef CAS PubMed.
- I. V. Astakhova, D. Lindegaard, A. D. Malakhov, V. A. Korshun and J. Wengel, Chem. Commun., 2010, 46, 8362 RSC; M. E. Østergaard and P. J. Hrdlicka, Chem. Soc. Rev., 2011, 40, 5771 RSC.
- K. K. Karlsen and J. Wengel, Nucleic Acid Ther., 2012, 22, 366 CAS.
- V. C. Spiteri and J. E. Moses, Angew. Chem., Int. Ed., 2010, 49, 31 CrossRef PubMed.
- I. K. Astakhova and J. Wengel, Chem.–Eur. J., 2013, 19, 1112 CrossRef CAS PubMed; M. M. Rubner, C. Holzhauser, P. R. Bohländer and H.-A. Wagenknecht, Chem.–Eur. J., 2012, 18, 1299 CrossRef PubMed; S. P. Sau and P. J. Hrdlicka, J. Org. Chem., 2012, 77, 5 CrossRef PubMed; A. H. El-Sagheer and T. Brown, Acc. Chem. Res., 2012, 45, 1258 CrossRef PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- A. Buhl, S. Page, N. H. H. Heegaard, P. von Landenberg and P. B. Luppa, Biosens. Bioelectron., 2009, 25, 198 CrossRef CAS PubMed.
- T. H. Winkler, S. Jahn and J. R. Kalden, Clin. Exp. Immunol., 1991, 85, 379 CrossRef CAS ; dsDNA-mAb32 and dsDNA-mAb33 correspond to clones 32.B9 and 33.H11, respectively.
- Y. Zhanga and X. Sun, Chem. Commun., 2011, 47, 3927 RSC.
- C.-H. Leung, D. S.-H. Chan, H.-Z. He, Z. Cheng, H. Yang and D.-L. Ma, Nucleic Acids Res., 2012, 40, 941 CrossRef CAS PubMed.
- L. Wang, Q. Zheng, Q. Zhang, H. Xu, J. Tong, C. Zhu and Y. Wan, Oncol. Lett., 2012, 4, 935 Search PubMed; S. Xie, Y. Chai, R. Yuan, L. Bai, Y. Yuan and Y. Wang, Anal. Chim. Acta, 2012, 755, 46 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of monomer M1, oligonucleotide synthesis and purification, click chemistry, NMR, MALDI-MS, IE HPLC and Tm data; fluorescence diagnostic assays and representative emission spectra, quantum yields, and molecular modeling. See DOI: 10.1039/c3cc45507f |
| This journal is © The Royal Society of Chemistry 2013 |