 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An RNA modification with remarkable resistance to RNase A†
Alice
Ghidini
a,
Charlotte
Ander
a,
Anna
Winqvist
b and
Roger
Strömberg
*a
aDepartment of Biosciences and Nutrition, Karolinska Institutet, Novum, Hälsovägen 7, 14183, Huddinge, Sweden. E-mail: Roger.Stromberg@ki.se
bMedivir AB, Lunastigen 7, 141 44, Huddinge, Sweden
First published on 8th August 2013
Abstract
A 3′-deoxy-3′-C-methylenephosphonate modified diribonucleotide is highly resistant to degradation by spleen phosphodiesterase and not cleaved at all by snake venom phosphodiesterase. The most remarkable finding is that, despite the fact that both the vicinal 2-hydroxy nucleophile and the 5′-oxyanion leaving group are intact, the 3′-methylenephosponate RNA modification is also highly resistant towards the action of RNase A.
Access to synthetic nucleic acid fragments has been pivotal in the development of life science research and modified oligonucleotides show considerable promise for disease therapeutics. A general theme in the different therapeutic approaches, such as siRNA1 (short interfering RNA), antisense technologies,2 including pre-mRNA splice-switching3 and even in the development of artificial ribonucleases,4–6 is the use of oligonucleotides that are stabilised towards enzymatic degradation and have an increased affinity for the target. However, due to the presumed lability in biological fluids, the oligoribonucleotide analogues developed typically do not carry a 2′-hydroxyl. The few examples include non-natural linkages not recognized by enzymes, such as internucleosidic amides7 and acetals.8 Among the most common modifications are instead 2′-O-alkyl groups9 and phosphorothioates,10 mostly without free 2′-hydroxyl groups. In addition, modified di- and oligonucleotides have been used as potential enzyme inhibitors and in investigations of enzymatic mechanisms for a number of decades, and the use has increased with the number of analogues available.11 An interesting modification that was introduced in diribonucleotides in 197012 was internucleoside 3′-deoxy-3′-C-methylenephosphonate linkage (Fig. 1). Later on, this was also incorporated in a trinucleotide13 but no data on stability to enzymatic cleavage have been presented.
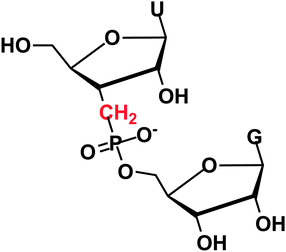 | ||
| Fig. 1 RNA with a 3′ methylenephosphonate linkage. | ||
The early synthesis work is difficult to utilize for making oligonucleotides and occurred before the development of the H-phosphonate approach14 and its application in oligonucleotide synthesis.15 After the latter appeared, it seemed likely that an analogous approach via alkylhydrogenphosphinates should be possible for incorporation of methylenephosphonate linkages into oligonucleotides. We have since developed synthetic methods for key steps towards ribonucleoside 3′-deoxy-3′-C-methylenephosphinates, i.e., a 3′-carbon extension at the nucleoside level,16 synthesis of methylenephosphinate building blocks17 and oxidation of methylenephosphinate linkages18 as well as evaluation of condensing agents for formation of the methylenephosphinate internucleoside linkage.19 The potential biological use of methylenephosphonate RNA is dependent on its stability towards nucleases and phosphodiesterases. Here we present a study on the stability of the methylenephosphonate linkage in a diribonucleotide and towards enzyme catalyzed degradation by two different phosphodiesterases/nucleases and RNase A.
The model dinucleotide, guanosine 5′-(uridine 3-deoxy-3′-C-methylenephosphonate) (UCH2pG, 1, Scheme 1) was synthesized and then fully deprotected and purified by HPLC (see ESI†). The UCH2pG dinucleotide was first subjected to snake venom phosphodiesterase (SVPD, PDE I) from Crotalus adamanteus and analysed by HPLC at different times. There was no cleavage observed with SVPD after two days of incubation (see ESI†), which is expected since the enzyme cleaves internucleosidic phosphateesters with the 3′-oxyanion as the leaving group, but in the modified dinucleotide this is replaced by a methylene group.
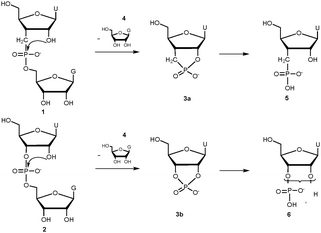 | ||
| Scheme 1 Pathways for the cleavage of UCH2pG and UpG dinucleotides with RNase A or base (monitoring is only for the first step since the ratio of dinucleotide to the sum of all products was evaluated). | ||
Both UCH2pG and the native diribonucleotide UpG were then subjected to spleen phosphodiesterase (PDE II), which is a 5′-exonuclease. HPLC analysis of the mixtures revealed that the methylenephosphonate analogue UCH2pG is considerably more stable than the native UpG (Fig. 2a), with about 75% remaining of the modified dimer under conditions where UpG is almost completely degraded.
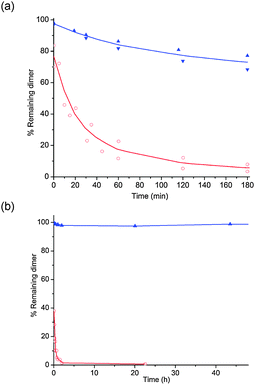 | ||
| Fig. 2 Graphs showing % remaining dinucleotide (UCH2pG in blue and UpG in red) at different times, when subjected to spleen exonuclease, PDE II (a) or RNase A (b) at 37 °C. Quantification of dinucleotide and product was done by integration of the RP-HPLC analysis of the mixtures. | ||
The complete resistance towards a 3′ exonuclease and high resistance towards a 5′-exonuclease suggest that the 3′-deoxy-3′-C-methylenephosphonate modification can be highly interesting for incorporation into therapeutic oligonucleotides. What then is the stability towards an RNA-degrading endonuclease?
When UCH2pG and the native diribonucleotide UpG were subjected to RNase A, the difference in rate of enzyme catalyzed cleavage was even more pronounced than with the spleen phosphodiesterase. HPLC analysis revealed that the methylenephosphonate analogue UCH2pG is hardly cleaved at all over two days and under conditions where the natural dimer UpG is almost completely degraded after a few minutes (Fig. 2b). In fact, even after seven days incubation with RNase A there was only a few percent cleavage of UCH2pG (see ESI†).
The methylenephosphonate modification clearly seems to be interesting for further exploration. But what also struck us is the quite remarkable resistance to the RNase A catalyzed cleavage, especially since the dinucleotide contains all groups suggested to interact in mechanisms for catalysis,20,21 including the 2′-hydroxy nucleophile, the 5′-oxyanion leaving group and a negatively charged phosphoryl functionality. The only difference between the substrates is the methylene group and the large effect on the rate of cleavage when they are catalyzed by the RNase A, which is not what can be expected by intrinsic chemical reactivity upon cyclization (Scheme 1). It is known that the rate of alkaline hydrolysis for cyclic phosphonateesters is only about a factor of two lower than for the corresponding cyclic phosphateesters.22 The ring strain effect23 makes cyclic esters somewhat different so, to make sure that there was no major intrinsic chemical difference between UCH2pG and UpG, we performed a study on the rate of hydrolysis under alkaline conditions (where a more or less concerted mechanism is expected).
Both dinucleotides were subjected to hydrolysis in 0.01 M and 0.05 M NaOH solutions (I = 0.2 M with NaCl) at 50 °C and monitored by RP-HPLC analysis at different times. The natural RNA dimer UpG was cleaved somewhat faster than the modified UCH2pG but the rate difference in both solutions is only a factor of about 1.7 (Fig. 3a). This clearly does not explain the large difference in rate found in the enzyme catalyzed cleavages, especially by RNase A. The methylenephosphonate RNA is interesting for incorporation in oligonucleotides but it is also intriguing why it is so stable towards RNase A catalyzed cleavage. Could the binding to the enzyme be hampered in some way?
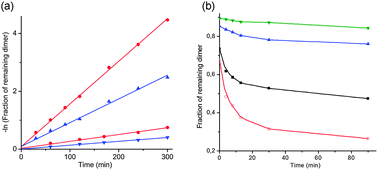 | ||
Fig. 3 (a) Graph showing the natural logarithm of the fraction of remaining dinucleotide (UpG in red and UCH2pG in blue) when subjected to hydrolysis in 0.01 M (lower two lines) and 0.05 M NaOH solutions (upper two lines) at 50 °C. (b) Graph showing % remaining UpG at different times, when subjected to RNase A, in the presence of different amounts of UCH2pG at 37 °C. (Red line: only UpG, i.e., 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0; black line: equimolar amounts, i.e., 1:1; blue line: 1:5; green line: 1:10.) :0; black line: equimolar amounts, i.e., 1:1; blue line: 1:5; green line: 1:10.) | ||
The methylene modification should stereoelectronically enforce a preference for the north (3′-endo) conformations of the sugar24,25 as it is found in a difluromethylenephosphonate,26 even though this has a more electron withdrawing group attached to the 3′-position. If the dinucleotide binding to RNase A would occur in another conformation, e.g., 2′-endo (south), one could expect that the overall rate would be higher with the native UpG that is less locked to a north conformation. If this were the reason for a much lower rate of cleavage with UCH2pG, one would not expect this compound to be able to compete efficiently with UpG in binding to the active site. If, however, the dimer binds in the north conformation one would expect UCH2pG to be a competitive inhibitor. UCH2pG is cleaved but at such a rate that it would essentially act as an inhibitor for the UpG substrate (at ten times higher concentration of RNase A the cleavage of UCH2pG is still slow but more prominent, see ESI†).
Competition experiments, where different amounts of UCH2pG were added to incubations of UpG with RNase A, were performed. It is evident that already at equimolar amounts of the two dinucleotides the rate of cleavage of UpG is substantially retarded (Fig. 3b, black line). As the amount of UCH2pG present is increased to 5 and 10 equivalents compared to UpG, the rate of cleavage of the natural dimer decreases even further (Fig. 3a, the two upper blue and green lines). This indicates that UCH2pG efficiently competes with UpG for the binding in the active site and the magnitude of the suppression even suggests that UCH2pG binds tighter than the native dimer. If the 3′-endo conformer is adopted upon binding, it would explain why UCH2pG is an efficient binding competitor since it is more locked in that conformation.
So why then is UCH2pG cleaved at such a low rate by RNase A? A couple of options involving a conformational change can be considered. The Breslow mechanism20 involves a phosphorane intermediate and if initial attack does not take place with the 5′-oxygen in an apical position, pseudorotation will be necessary for completion of the reaction. This would be severely retarded since it would involve an apical methylene substituent that has a very low apicophilicity.27 Another option is that the initially bound 3′-endo conformer has to flip to 2′-endo before the attack on phosphorus can take place, and this is retarded by the conformational preference of the methylenephosphonate, perhaps accentuated when bound to the enzyme. It is also plausible that lysine-41, suggested to stabilize charge build up on the non-bridging phosphoryl oxygens,28 is somehow involved and interacts with the 3′-oxygen. This could also be connected to one of the conformational changes above.
In view of what is known about the RNase A mechanism it is surprising that the methylenephosphonate dinucleotide is highly resistant to RNase A catalyzed cleavage. It is also interesting that there still seem to be details in the mechanism of catalysis for RNase A that are not yet resolved and hopefully the methylenephosphonate modification can contribute to further studies, e.g., by co-crystallization with the enzyme and further enzymology.
That the modification leads to a higher resistance towards degradation by both 3′-exo and 5′-exonucleases as well as an RNase endonuclease activity makes methylenephosphonate RNA interesting for incorporation into therapeutic oligonucleotides. In particular, it would be interesting to evaluate methylenephosphonate RNA in the antisense strand of therapeutic siRNAs.
We gratefully acknowledge financial support from The Swedish Research Council and EU Marie Curie network funding (EC-FP7-ITN-2008-238679).
Notes and References
- Y. Dorsett and T. Tuschl, Nat. Rev. Drug Discovery, 2004, 3, 318 CrossRef CAS PubMed; M. Manoharan, Curr. Opin. Chem. Biol., 2004, 8, 570 CrossRef PubMed.
- S. T. Crooke, Annu. Rev. Med., 2004, 55, 61 CrossRef CAS PubMed; S. T. Crooke, Curr. Mol. Med., 2004, 4, 465 CrossRef.
- J. Bauman, N. Jearawiriyapaisarn and R. Kole, Oligonucleotides, 2009, 19, 1 CrossRef CAS PubMed.
- J. R. Morrow and O. Iranzo, Curr. Opin. Chem. Biol., 2004, 8, 192 CrossRef CAS PubMed.
- H. Åström, N. H. Williams and R. Strömberg, Org. Biomol. Chem., 2003, 1461 Search PubMed; H. Åström and R. Strömberg, Org. Biomol. Chem., 2004, 2, 1901 Search PubMed.
- M. Murtola and R. Strömberg, Org. Biomol. Chem., 2008, 6, 3837 Search PubMed; M. Murtola, M. Wenska and R. Strömberg, J. Am. Chem. Soc., 2010, 132, 8984 CrossRef CAS PubMed.
- E. Rozners, D. Katkevica, E. Bizdena and R. Strömberg, J. Am. Chem. Soc., 2003, 125, 12125 CrossRef CAS PubMed.
- E. Rozners and R. Strömberg, J. Org. Chem., 1997, 62, 1846 CrossRef CAS.
- S. M. Freier and K. H. Altmann, Nucleic Acids Res., 1997, 25, 4429 CrossRef CAS PubMed.
- R. S. Geary, R. Z. Yu, A. Siwkowski and A. Levin, Antisense Drug Technology, 2nd edn, 2008, 305 Search PubMed; P. Guga and M. Koziolkiewicz, Chem. Biol. Artif. Nucleic Acids, 2012, 259 Search PubMed; H. Almer, J. Stawinski and R. Strömberg, Nucleic Acids Res., 1996, 24, 3811 CrossRef CAS PubMed; H. Almer and R. Strömberg, J. Am. Chem. Soc., 1996, 118, 7921 CrossRef.
- M. Warashina, Y. Takagi, W. J. Stec and K. Taira, Curr. Opin. Biotechnol., 2000, 11, 354 CrossRef CAS.
- H. P. Albrecht, G. H. Jones and J. G. Moffatt, J. Am. Chem. Soc., 1970, 92, 5511 CrossRef CAS.
- A. Mazur, B. E. Tropp and R. Engel, Tetrahedron, 1984, 40, 3949 CrossRef CAS.
- P. J. Garegg, T. Regberg, J. Stawinski and R. Strömberg, Chem. Scr., 1985, 25, 280–282 Search PubMed; P. J. Garegg, T. Regberg, J. Stawinski and R. Strömberg, Chem. Scr., 1986, 26, 59 Search PubMed; T. Regberg, J. Stawinski and R. Strömberg, Nucleosides Nucleotides, 1988, 7, 23 Search PubMed; S. Sigurdsson and R. Strömberg, J. Chem. Soc., Perkin Trans. 2, 2002, 1682 RSC.
- P. J. Garegg, C. Henrichson, I. Lindh, T. Regberg, J. Stawinski and R. Strömberg, Tetrahedron Lett., 1986, 27, 4051 CrossRef CAS; B. C. Froehler and M. D. Matteucci, Tetrahedron. Lett., 1986, 27, 469 CrossRef; B. C. Froehler, P. G. Ng and M. D Matteucci, Nucleic Acids Res., 1986, 14, 5399 CrossRef PubMed; P. J. Garegg, C. Henrichson, I. Lindh, T. Regberg, J. Stawinski and R. Strömberg, Tetrahedron Lett., 1986, 27, 4055 CrossRef.
- A. Winqvist and R. Strömberg, Eur. J. Org. Chem., 2001, 4305 CrossRef CAS.
- A. Winqvist and R. Strömberg, Eur. J. Org. Chem., 2002, 1509 CrossRef CAS.
- A. Winqvist and R. Strömberg, Eur. J. Org. Chem., 2002, 3140 CrossRef CAS.
- A. Winqvist and R. Strömberg, Eur. J. Org. Chem., 2008, 1705 CrossRef CAS.
- R. Breslow and W. H. Chapman Jr, Proc. Natl. Acad. Sci. U. S. A., 1996, 10018 CrossRef CAS.
- C. M. Cuchillo, M. V. Nogues and R. T. Raines, Biochemistry, 2011, 50, 7835 CrossRef CAS PubMed; R. T. Raines, Chem. Rev., 1998, 98, 1045 CrossRef PubMed.
- R. Kluger and S. D. Taylor, J. Am. Chem. Soc., 1990, 112, 6669 CrossRef CAS.
- A. Eberhard and F. H. Westheimer, J. Am. Chem. Soc., 1965, 87, 253 CrossRef CAS; R. Kluger and S. D. Taylor, J. Am. Chem. Soc., 1991, 113, 5714 CrossRef.
- Use of NMR for conformational analysis (see ref. 26) was attempted but we were unable to resolve the coupling constant for coupling between 1′-H and 2′-H. That this coupling is so small is in itself an indication of a high preference for the north conformers.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333 CrossRef CAS.
- C. Fressigne, S. Piettre, E. Condamine, C. Altona and A. Gautier, Tetrahedron, 2005, 61, 4769 CrossRef CAS PubMed.
- R. R. Holmes, J. Am. Chem. Soc., 1978, 100, 433 CrossRef CAS; R. K. Oram and S. Trippett, J. Chem. Soc., Chem. Commun., 1972, 553 Search PubMed.
- J. M. Messmore, D. N. Fuchs and R. T. Raines, J. Am. Chem. Soc., 1995, 117, 8057 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc44621b |
| This journal is © The Royal Society of Chemistry 2013 |