 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed intermolecular C–H amination of simple hydrocarbons using the shelf-stable nonafluorobutanesulfonyl azide†
José Ramón Suárez* and
Jose Luis Chiara*
Instituto de Química Orgánica General, CSIC, Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: jl.chiara@csic.es; Fax: +34-915644853; Tel: +34-915622900
First published on 13th August 2013
Abstract
A new procedure has been developed for the direct intermolecular C–H amination of simple hydrocarbons using shelf-stable nonafluorobutanesulfonyl azide in the presence of a dirhodium(II) tetracarboxylate catalyst under mild reaction conditions. Some mechanistic details are briefly discussed on the basis of control experiments.
The direct chemoselective transformation of unactivated sp3-hybridized C–H bonds into C–N bonds has emerged as a powerful tool in organic chemistry and the chemical industry that can streamline chemical synthesis by saving functional group transformations (such as hydroxyl to amine) and protection–deprotection steps.1 Typical aminating reagents used for this purpose are high-energy species bearing electron-deficient N-substituents and capable of generating multiple bonds between late transition metals and nitrogen, such as iminoiodinanes (often prepared in situ), N-haloamines, and azides. Intra- and intermolecular protocols have been described based on Mn,2 Fe,2c,3 Ru,2b,4 Co,2c,5 Rh,3a,6 Ir,4b,7 Ni,2c Pd,8 Cu,1c,9 Ag,10 Au,11 and Zn12 catalysts, including asymmetric versions,2b,4b,6d,f,7 as well as under metal-free13,14 conditions. In most of these transformations the reactive species is believed to be a (metal-)nitrene intermediate, which regioselectively inserts into the most electron-rich C–H bond unless steric factors contravene.
The use of azides is particularly appealing in this context since the reaction is highly atom-efficient and environmentally friendly, producing only gaseous nitrogen as a by-product and not requiring the addition of an oxidant. Polyfluoroalkanesulfonyl azides are the most electrophilic among organic azides, but have been very scarcely used in amination reactions13e,15 probably due to the highly hazardous nature of their simplest and most typically employed representative, triflyl azide (TfN3).16 However, it has been recently shown that the higher molecular weight analog nonafluorobutanesulfonyl azide (NfN3) is a more efficient, shelf-stable and economic reagent in diazo-transfer reactions17 and other useful transformation mediated by TfN3.17f,18 In connection with our recent studies on new synthetic applications of NfN3,17b–d,f here we describe how this reagent can perform the C–H amination of simple hydrocarbons under mild thermal conditions in the presence of a metal catalyst.
We selected indane as a model substrate for an initial screening of reaction conditions using 1.2 equivalents of NfN3 (Table 1). No reaction occurred under simple thermal conditions in 1,2-dichloroethane (DCE) at 90–110 °C (below the reported decomposition temperature of NfN3 of ca. 120 °C)13e for 12 hours in the absence of a metal catalyst (entry 1). After testing several transition metal salts (5 mol%) (entries 2–5), we found that dirhodium(II) tetracarboxylates promoted the reaction smoothly to afford the nonafluorobutanesulfonamide 1 in good isolated yields (entries 6, 12 and 13). The amination took place exclusively at the benzylic carbon. Only a marginal improvement in product yield was obtained with the more active Rh2(esp)2 catalyst (entry 13).6c A decrease in the reaction temperature had a dramatic deleterious effect, the reaction becoming sluggish below 70 °C (entries 7 and 8). Catalyst loadings as low as 1 mol% could be employed at the expense of a reduced, but still synthetically useful, amination yield (entry 10). Use of an excess of NfN3 (2 equivalents) resulted in a slightly reduced product yield without formation of polyaminated products or imine5a,9c (entry 11). Optimum results were obtained at 90 °C employing a 3–5 mol% of catalyst and 1.2 equivalents of NfN3 (entries 6, 9, 12 and 13).
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Temp (°C) | Yielda (%) |
| a Isolated yield.b No reaction.c Two equivalents of NfN3 were used. | |||
| 1 | None | 90–110 | n.r.b |
| 2 | Cu(OTf)2 (5) | 90 | n.r. |
| 3 | CuBr (5) | 90 | n.r. |
| 4 | Pd(OAc)2 (5) | 90 | n.r. |
| 5 | AgOTf (5) | 90 | n.r. |
| 6 | Rh2(OAc)4 (5) | 90 | 83 |
| 7 | Rh2(OAc)4 (5) | 50 | <5 |
| 8 | Rh2(OAc)4 (5) | 70 | 20 |
| 9 | Rh2(OAc)4 (3) | 90 | 82 |
| 10 | Rh2(OAc)4 (1) | 90 | 63 |
| 11c | Rh2(OAc)4 (5) | 90 | 75 |
| 12 | Rh2(O2CC7H15)4 (5) | 90 | 80 |
| 13 | Rh2(esp)2 (5) | 90 | 84 |

We next examined the substrate scope under the cost-efficient optimized conditions using 5 mol% Rh2(OAc)4 at 90 °C in DCE. Simple aromatic compounds bearing primary, secondary or tertiary benzylic hydrogens where regioselectively monosulfamidated at the benzylic position with moderate to good isolated yields (47–80%) (Scheme 1). Amination of the aromatic ring was never observed under the employed experimental conditions.13a,15a,19 In the case of isochromane, the reaction occurred exclusively at the ethereal benzylic carbon (compound 3). However, in contrast to related Rh(II)-catalyzed amination reactions of aliphatic C–H bonds with aryl azides,6i no reaction was observed for substrates bearing carbonyl, ester, carbamoyl or sulfonamido groups, the starting compound being recovered unchanged after heating at 90 °C for 16 h (Scheme 1, last row).
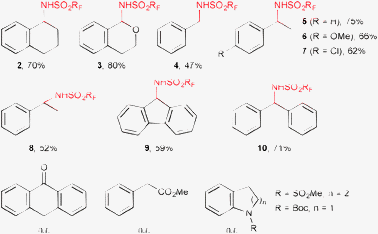 | ||
| Scheme 1 Intermolecular amination of benzylic C–H bonds. | ||
Encouraged by these results, we next examined the scope of the transformation on simple hydrocarbon substrates bearing no functional groups (Scheme 2). Moderate to good isolated yields (45–70%) of monosulfamidated products were obtained under the previous optimized conditions using the hydrocarbons as limiting reagents (14–18) or as solvents (11–13). In the case of adamantane, amination took place regioselectively at the tertiary C–H bond, with no product arising from methylene amination being detected in spite of the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 statistical preference for insertion at the methylene C–H bonds in this molecule. In contrast, trans-decalin was aminated exclusively at the methylene carbons, yielding an inseparable 2:3 mixture (determined from the 1H NMR of the crude) of two regio- or stereoisomers (15). The larger steric encumbrance of the axially oriented methine hydrogens probably explains this selectivity. Thus, in cis-decalin an inseparable mixture of three regio- and/or stereo-isomeric sulfonamides (16) was formed showing that amination at the now more accessible methine carbon has taken place. The strong signal overlap observed in the 1H NMR of the decalin reaction products impeded their stereochemical assignment.9f A similar change in 2°–3° site selectivity was observed for cis- and trans-1,4-dimethylcyclohexane. While the cis-isomer reacted preferentially at the methylene carbons to afford an inseparable 6.7:1 mixture of 2°–3° sulfonamides 17 as single (unassigned) diastereoisomers, the trans-isomer gave a 2.2:3.1 mixture of 2°(2 diastereoisomers)–3°(single diastereoisomer) sulfonamides 18, respectively, in spite of the 4:1 statistical preference for insertion at the methylene C–H bonds in this molecule, indicating again the preferential amination of the more accessible equatorially oriented methine C–H. We next studied the regioselectivity of the reaction in isobutylbenzene and 1-ethyl-4-methylbenzene (Scheme 3). The observed reactivity ratio for amination at benzylic vs. tertiary alkylic and at primary vs. secondary benzylic C–H bonds were, respectively, 2:1 and 4.2:1 (statistically corrected), which follow the trend of their expected relative C–H bond dissociation energies [BDE(Me3C–H) = 96.5 kcal mol−1; BDE(PhCH2–H) = 89.8 kcal mol−1; BDE(PhCH(Me)–H) = 85.4 kcal mol−1].20
:1 statistical preference for insertion at the methylene C–H bonds in this molecule. In contrast, trans-decalin was aminated exclusively at the methylene carbons, yielding an inseparable 2:3 mixture (determined from the 1H NMR of the crude) of two regio- or stereoisomers (15). The larger steric encumbrance of the axially oriented methine hydrogens probably explains this selectivity. Thus, in cis-decalin an inseparable mixture of three regio- and/or stereo-isomeric sulfonamides (16) was formed showing that amination at the now more accessible methine carbon has taken place. The strong signal overlap observed in the 1H NMR of the decalin reaction products impeded their stereochemical assignment.9f A similar change in 2°–3° site selectivity was observed for cis- and trans-1,4-dimethylcyclohexane. While the cis-isomer reacted preferentially at the methylene carbons to afford an inseparable 6.7:1 mixture of 2°–3° sulfonamides 17 as single (unassigned) diastereoisomers, the trans-isomer gave a 2.2:3.1 mixture of 2°(2 diastereoisomers)–3°(single diastereoisomer) sulfonamides 18, respectively, in spite of the 4:1 statistical preference for insertion at the methylene C–H bonds in this molecule, indicating again the preferential amination of the more accessible equatorially oriented methine C–H. We next studied the regioselectivity of the reaction in isobutylbenzene and 1-ethyl-4-methylbenzene (Scheme 3). The observed reactivity ratio for amination at benzylic vs. tertiary alkylic and at primary vs. secondary benzylic C–H bonds were, respectively, 2:1 and 4.2:1 (statistically corrected), which follow the trend of their expected relative C–H bond dissociation energies [BDE(Me3C–H) = 96.5 kcal mol−1; BDE(PhCH2–H) = 89.8 kcal mol−1; BDE(PhCH(Me)–H) = 85.4 kcal mol−1].20

 | ||
| Scheme 3 Competitive benzylic vs. tertiary alkylic and primary vs. secondary C–H bond amination. | ||
A modest primary kinetic isotope effect (KIE, kH/kD = 2.47 at 90 °C; see ESI†) was measured for methylene C–H bond amination in a competition reaction using a 1:1 mixture of cyclohexane and cyclohexane-d12 and a limiting amount of NfN3. This value is considerably lower than those previously reported for reactions proceeding via a triplet nitrene C–H abstraction/radical rebound amination mechanism (kH/kD > 5–6),1a,4a but it is in the same range as those evaluated for analogous rhodium-catalyzed C–H aminations (KIE = 1.2–2.6)1d proposed to proceed via concerted asynchronous singlet nitrene insertion. In line with this observation is that the tertiary sulfonamides 17c and 18c were diastereoisomerically different (see ESI†)14d suggesting that the amination took place with retention of configuration, which can rule out a radical stepwise mechanism. Based on these results, a concerted asynchronous pathway can be reasonably proposed for the present nitrene C–H insertion with only partial C–H bond breaking in the transition state.
The sulfonamide products could be further transformed taking advantage of the known reactivity of polyfluoroalkanesulfonamides.21 Thus, N-alkylation could be readily performed by reaction of the sulfonamide anion with an alkyl halide (Scheme 4). The resultant N,N-disubstituted nonafluorobutanesulfonamide showed restricted rotation around the S–N bond, which caused a considerable broadening of the 1H NMR signals at room temperature.22 Subsequent treatment with Red-Al in toluene under thermal conditions afforded the corresponding N-alkylated amine in very good yield. This efficient two-step process can allow simple access to a variety of secondary amines, thus widely expanding the synthetic potential of the present C–H bond amination approach.
 | ||
| Scheme 4 N-alkylation/reductive deprotection of sulfonamides. | ||
In summary, we have reported a new intermolecular C(sp3)–H amination of simple hydrocarbons using the shelf-stable nonafluorobutanesulfonyl azide in the presence of a dirhodium(II) tetracarboxylate catalyst. The amination products were obtained in moderate to good yields and could be further transformed to secondary amines via a simple two-step process. Possible mechanistic pathways for the amination process were briefly discussed on the basis of control experiments.
We gratefully acknowledge support by the Spanish Ministerio de Ciencia e Innovación (project MAT2010-20646-C04-03), the European Social Fund and Comunidad de Madrid (project S2009/PPQ-1634 “AVANCAT”), and CSIC for a JAEDOC contract to J.R.S. Thanks are due also to Prof. Jesús Sanz (IQOG-CSIC) for his valuable assistance with the KIE experiments.
Notes and references
- (a) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061–5074 RSC; (b) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831–3846 RSC; (c) R. T. Gephart and T. H. Warren, Organometallics, 2012, 31, 7728–7752 CrossRef CAS; (d) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed.
- (a) R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400–1401 RSC; (b) J.-L. Liang, J.-S. Huang, X.-Q. Yu, N. Zhu and C.-M. Che, Chem.–Eur. J., 2002, 8, 1563–1572 CrossRef CAS; (c) S. Liang and M. P. Jensen, Organometallics, 2012, 31, 8055–8058 CrossRef CAS.
- (a) R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728–6729 CrossRef CAS; (b) Z. Wang, Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2008, 10, 1863–1866 CrossRef CAS PubMed; (c) Y. Liu and C.-M. Che, Chem.–Eur. J., 2010, 16, 10494–10501 CrossRef CAS PubMed; (d) L. Liang, H. Lv, Y. Yu, P. Wang and J.-L. Zhang, Dalton Trans., 2012, 41, 1457–1460 RSC; (e) S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036–2039 CrossRef CAS PubMed; (f) E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed.
- (a) S. K.-Y. Leung, W.-M. Tsui, J.-S. Huang, C.-M. Che, J.-L. Liang and N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629–16640 CrossRef CAS PubMed; (b) Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739–1742 CrossRef CAS PubMed.
- (a) J. D. Harden, J. V. Ruppel, G.-Y. Gao and X. P. Zhang, Chem. Commun., 2007, 4644–4646 RSC; (b) H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192–10196 CrossRef CAS PubMed; (c) Y.-H. Ye, J. Zhang, G. Wang, S.-Y. Chen and X.-Q. Yu, Tetrahedron, 2011, 67, 4649–4654 CrossRef CAS PubMed.
- (a) I. Nägeli, C. Baud, G. Bernardinelli, Y. Jacquier, M. Moraon and P. Müllet, Helv. Chim. Acta, 1997, 80, 1087–1105 CrossRef; (b) C. G. Espino and J. Du Bois, Angew. Chem., Int. Ed., 2001, 40, 598–600 CrossRef CAS; (c) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef CAS PubMed; (d) R. P. Reddy and H. M. L. Davies, Org. Lett., 2006, 8, 5013–5016 CrossRef CAS PubMed; (e) K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562–568 CrossRef CAS PubMed; (f) C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2007, 130, 343–350 CrossRef PubMed; (g) K. Huard and H. Lebel, Chem.–Eur. J., 2008, 14, 6222–6230 CrossRef CAS PubMed; (h) C. Lescot, B. Darses, F. Collet, P. Retailleau and P. Dauban, J. Org. Chem., 2012, 77, 7232–7240 CrossRef CAS PubMed; (i) Q. Nguyen, K. Sun and T. G. Driver, J. Am. Chem. Soc., 2012, 134, 7262–7265 CrossRef CAS PubMed; (j) A. Nörder, S. A. Warren, E. Herdtweck, S. M. Huber and T. Bach, J. Am. Chem. Soc., 2012, 134, 13524–13531 CrossRef PubMed.
- M. Ichinose, H. Suematsu, Y. Yasutomi, Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2011, 50, 9884–9887 CrossRef CAS PubMed.
- (a) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2011, 134, 3–6 CrossRef PubMed; (b) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2011, 134, 7–10 CrossRef PubMed.
- (a) M. M. Díaz-Requejo, T. R. Belderraín, M. C. Nicasio, S. Trofimenko and P. J. Pérez, J. Am. Chem. Soc., 2003, 125, 12078–12079 CrossRef PubMed; (b) M. R. Fructos, S. Trofimenko, M. M. Díaz-Requejo and P. J. Pérez, J. Am. Chem. Soc., 2006, 128, 11784–11791 CrossRef CAS PubMed; (c) Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961–9964 CrossRef CAS PubMed; (d) D. A. Powell and H. Fan, J. Org. Chem., 2010, 75, 2726–2729 CrossRef CAS PubMed; (e) R. T. Gephart, D. L. Huang, M. J. B. Aguila, G. Schmidt, A. Shahu and T. H. Warren, Angew. Chem., Int. Ed., 2012, 51, 6488–6492 CrossRef CAS PubMed; (f) Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547–2550 CrossRef CAS PubMed; (g) Z. Ni, Q. Zhang, T. Xiong, Y. Zheng, Y. Li, H. Zhang, J. Zhang and Q. Liu, Angew. Chem., Int. Ed., 2012, 51, 1244–1247 CrossRef CAS PubMed.
- (a) B. P. Gómez-Emeterio, J. Urbano, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2008, 27, 4126–4130 CrossRef; (b) Á. Beltrán, C. Lescot, M. M. Díaz-Requejo, P. J. Pérez and P. Dauban, Tetrahedron, 2013, 69, 4488–4492 CrossRef PubMed.
- (a) Z. Li, D. A. Capretto, R. O. Rahaman and C. He, J. Am. Chem. Soc., 2007, 129, 12058–12059 CrossRef CAS PubMed; (b) Y. Zhang, B. Feng and C. Zhu, Org. Biomol. Chem., 2012, 10, 9137–9141 RSC.
- B. Kalita, A. A. Lamar and K. M. Nicholas, Chem. Commun., 2008, 4291–4293 RSC.
- For pioneering work, see: (a) D. S. Breslow, M. F. Sloan, N. R. Newburg and W. B. Renfrow, J. Am. Chem. Soc., 1969, 91, 2273–2279 CrossRef CAS; (b) T. Shingaki, M. Inagaki, N. Torimoto and M. Takebayashi, Chem. Lett., 1972, 1181–1184 CrossRef CAS; (c) D. S. Breslow, E. I. Edwards, E. C. Linsay and H. Omura, J. Am. Chem. Soc., 1976, 98, 4268–4275 CrossRef CAS; (d) N. Torimoto, T. Shingaki and T. Nagai, J. Org. Chem., 1978, 43, 631–633 CrossRef CAS; (e) S.-Z. Zhu, J. Chem. Soc., Perkin Trans. 1, 1994, 2077–2081 RSC.
- (a) H. F. Bettinger, M. Filthaus, H. Bornemann and I. M. Oppel, Angew. Chem., Int. Ed., 2008, 47, 4744–4747 CrossRef CAS PubMed; (b) A. A. Lamar and K. M. Nicholas, J. Org. Chem., 2010, 75, 7644–7650 CrossRef CAS PubMed; (c) H. J. Kim, J. Kim, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2011, 133, 16382–16385 CrossRef CAS PubMed; (d) M. Ochiai, K. Miyamoto, T. Kaneaki, S. Hayashi and W. Nakanishi, Science, 2011, 332, 448–451 CrossRef CAS PubMed; (e) Q. Xue, J. Xie, H. Li, Y. Cheng and C. Zhu, Chem. Commun., 2013, 49, 3700–3702 RSC.
- (a) N. Kamigata, K. Yamamoto, O. Kawakita, K. Hikita, H. Matsuyama, M. Yoshida and M. Kobayashi, Bull. Chem. Soc. Jpn., 1984, 57, 3601–3602 CrossRef CAS; (b) L. Benati, D. Nanni and P. Spagnolo, J. Org. Chem., 1999, 64, 5132–5138 CrossRef CAS.
- B. A. Shainyan and L. L. Tolstikova, Chem. Rev., 2013, 113, 699–733 CrossRef CAS PubMed.
- (a) S. Yekta, V. Prisyazhnyuk and H.-U. Reissig, Synlett, 2007, 2069–2072 CAS; (b) J. R. Suárez, B. Trastoy, M. E. Pérez-Ojeda, R. Marin-Barrios and J. L. Chiara, Adv. Synth. Catal., 2010, 352, 2515–2520 CrossRef; (c) B. Trastoy, M. E. Pérez-Ojeda, R. Sastre and J. L. Chiara, Chem.–Eur. J., 2010, 16, 3833–3841 CrossRef CAS PubMed; (d) J. L. Chiara and J. R. Suárez, Adv. Synth. Catal., 2011, 353, 575–579 CrossRef CAS; (e) J. Gu, W. Xiong, Z. Zhang and S. Zhu, Synthesis, 2011, 1717–1722 CAS; (f) J. R. Suárez, J. Kwiczak, K. Grenda, M. L. Jimeno and J. L. Chiara, Adv. Synth. Catal., 2013, 355, 913–918 CrossRef.
- (a) W. Xiong, Y. Xin, J. Han and S. Zhu, J. Fluorine Chem., 2010, 131, 867–872 CrossRef CAS PubMed; (b) W. Xiong, Y. Xin, J. Zhao and S. Zhu, Synthesis, 2011, 1142–1148 CAS; (c) W. Xiong, H. Zhang, Y. Xin and S. Zhu, Tetrahedron, 2011, 67, 2232–2237 CrossRef CAS PubMed; (d) W.-t. Xiong, J.-w. Zhao, J.-w. Gu and S. Zhu, Tetrahedron, 2011, 67, 5235–5243 CrossRef CAS PubMed.
- S. Zhu and P. He, Tetrahedron, 2005, 61, 5679–5685 CrossRef CAS PubMed.
- (a) D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493–532 CrossRef CAS; (b) S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- (a) J. B. Hendrickson, R. Bergeron and D. D. Sternbach, Tetrahedron, 1975, 31, 2517–2521 CrossRef CAS; (b) K. Miyamoto, M. M. Hoque and S. Ogasa, J. Org. Chem., 2012, 77, 8317–8320 CrossRef CAS PubMed.
- I. M. Lyapkalo, H.-U. Reissig, A. Schäfer and A. Wagner, Helv. Chim. Acta, 2002, 85, 4206–4215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and copies of 1H, 19F, and 13C NMR spectra. See DOI: 10.1039/c3cc44594a |
| This journal is © The Royal Society of Chemistry 2013 |