 Open Access Article
Open Access ArticleTandem thia-Fries rearrangement – cyclisation of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate benzyne precursors†‡
Catherine
Hall
,
Jaclyn L.
Henderson
,
Guillaume
Ernouf
and
Michael F.
Greaney
*
School of Chemistry, University of Manchester, Oxford Rd, Manchester M13 9PL, UK. E-mail: michael.greaney@manchester.ac.uk
First published on 9th July 2013
Abstract
A novel transformation of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate aryne precursors is described.
The introduction of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate as a precursor to ortho-benzyne has fueled a renaissance in aryne chemistry.1 Discovered in 1983 by Kobayashi,2 the precursor generates benzyne under the notably mild conditions of simple fluoride treatment at room temperature. The initial conditions of CsF in MeCN have been expanded to include a variety of fluoride sources and solvent combinations, conferring a degree of control on the rate of benzyne generation and thus opening up new areas of chemistry that were previously inaccessible using classical aryne precursors.
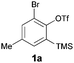
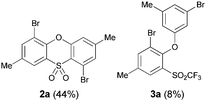
In the course of developing new transformations of ortho-benzyne,3 we observed that fluoride treatment of bromo-substituted precursor 1a led to the unexpected formation of phenoxathiin-dioxide 2a in 51% yield (Scheme 1). The heterocycle was isolated as a single regio-isomer and characterised by single crystal X-ray diffraction4 (Fig. 1). This intriguing reaction is without precedent in aryne chemistry, prompting us to conduct further investigations to try and understand the mechanistic pathway leading to 2a. In addition, phenoxathiin-dioxides are a scarcely studied class of heterocycle, and novel methods for their synthesis could prove useful.5
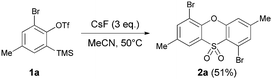 | ||
| Scheme 1 Phenoxathiin-dioxide synthesis. | ||
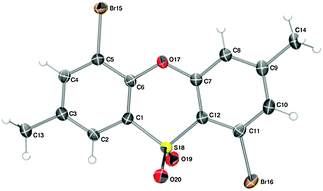 | ||
| Fig. 1 X-ray structure of 2a. Thermal ellipsoids at 50% probability. | ||
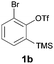
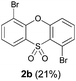
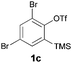
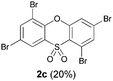
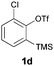
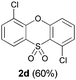
We began our study by repeating the reaction at room temperature, observing the formation of 2a in slightly lower yield along with a small amount of the triflone 3a (Table 1, entry 1). Isolation of 3a is significant as it suggests that it, or a related species, may be a precursor to the final phenoxathiin-dioxide product 2a. The methyl group in 1a was not a requirement for reaction (entry 2), with the bromo-compound 1b providing 2b in 21% yield at room temperature along with small amounts of uncyclised triflone. The yield of the process could be improved by raising the temperature to 60 °C (47% yield, entry 3), or by adding toluene to the solvent system, with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN:toluene mix providing 2b in 60% yield at room temperature (entry 4). Addition of a less polar solvent would be expected to slow the rate of reaction of CsF with the silane starting material 1a. Adding an additional bromine atom to the substrate reduced the reaction efficiency (entry 5), but the chloro derivative 1c proved successful in the reaction, providing 2d in 60% yield (entry 6).
:1 MeCN:toluene mix providing 2b in 60% yield at room temperature (entry 4). Addition of a less polar solvent would be expected to slow the rate of reaction of CsF with the silane starting material 1a. Adding an additional bromine atom to the substrate reduced the reaction efficiency (entry 5), but the chloro derivative 1c proved successful in the reaction, providing 2d in 60% yield (entry 6).
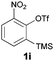
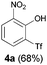
Moving away from halogens at the ortho-position to the trifluoromethanesulfonate, however, immediately shut down the reaction. Phenyl, methoxy, methyl and simple hydrogen-substitution all gave intractable mixtures from which no phenoxathiin dioxide could be isolated (entry 7). The ortho-nitro substrate 1i, however, reacted to give the rearranged phenol 4a in 68% yield (entry 8).
Collectively, these results point to a mechanism involving an anionic thia-Fries rearrangement. Fluoride treatment of 1 results in C–Si bond cleavage as the first step in the mechanism, followed by thia-Fries reaction of the resulting anion.6 The phenolate 6 can then react with an aryne molecule 7, formed from 5via trifluoromethanesulfonate elimination in the established aryne generation pathway. Cyclisation of the resultant phenyl anion 8 onto the trifluoromethanesulfonate group gives the heterocyclic phenoxathiin-dioxide products (Scheme 2).
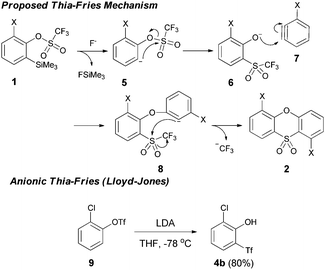 | ||
| Scheme 2 Proposed mechanism for phenoxathiin-dioxide formation. | ||
The anionic thia-Fries rearrangement was first reported by Lloyd-Jones and co-workers (also working in the context of aryne generation).7 Treatment of 2-(chloro)phenyl trifluoromethanesulfonate 9 with LDA at −78 °C led to either thia-Fries rearrangement (to compound 4b) or aryne generation, a dichotomy that could be controlled by the amount of DIPA present in the reaction. Subsequent elucidation of the reaction mechanism revealed the degree of metalation to be the critical factor in partitioning the reaction between thia-Fries and aryne generation.6 The reaction has also been demonstrated to occur for 2-(trimethylsilyl)phenyl trifluoromethanesulfonates, with Butenschön and co-workers reporting examples using aryltricarbonylchromium complexes as substrates.8,9 Our substrate scope in Table 1 is in line with literature reports on the thia-Fries rearrangement of trifluoromethanesulfonates. Electron withdrawing groups ortho to the trifluoromethanesulfonate, halogens in particular, activate this pathway and subsequent phenoxathiin-dioxide formation. Moving away from this substitution pattern results in the reaction shutting down, in line with literature precedent. The nitro-containing substrate 1i clearly demonstrates the thia-Fries pathway in operation, providing the rearranged phenol 4a as the only product.9a It appears that the strongly electron withdrawing nitro group deactivates 1i to benzyne formation, providing no aryne 7 for further reaction to the heterocyclic products 2. Indeed, we were unable to find any reports of 6-nitro-2-(trimethylsilyl)phenyl trifluoromethanesulfonates being successfully used as aryne precursors in the literature. Halo-substituted compounds, however, can generate arynes and undergo further reaction to give the adduct 8. Protonation of this anion affords the triflones 3 isolated as side-products. In contrast to Lloyd-Jones' reaction system in the cryogenic regime, where thia-Fries rearrangement was orthogonal to aryne generation depending on reaction conditions, here we require the substrate 1 to bifurcate between both thia-Fries and aryne generation in the same reaction. The final cyclisation of anion 8 involves displacement of CF3 anion from the triflone, a process precedented for Grignard addition in sulfone synthesis.9b,10
Further experiments were carried out to test this mechanistic conjecture. We prepared the halo-phenol 4b separately and reacted it with benzyne under the reaction conditions. Successful cyclisation to the phenoxathiin-dioxide product 2f was observed, (along with formation of 3c), establishing phenol-aryne addition as a viable step in the mechanism (Scheme 3). Re-subjection of triflones such as 3c to the reaction conditions did not result in cyclisation to the heterocyclic products 2. This indicates that the neutral molecule is insufficiently nucleophilic to undergo intramolecular attack at the triflone group, and reaction through the anionic intermediate 8 is necessary to displace CF3 at the sulfur centre. The formation of small amounts of 3 in the reaction is likely due to competitive protonation from the MeCN solvent, a common side-product in aryne couplings involving silyl-trifluoromethanesulfonate precursors 1.3e
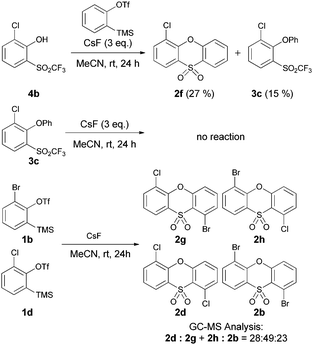 | ||
| Scheme 3 Mechanistic studies. | ||
Finally, we conducted cross-over experiments with two different aryne precursors in the same pot, 1b and 1d. A statistical mixture of hetero- and homo-coupled phenoxathiin-dioxides were observed on GC-MS analysis, supporting the dual reaction mode of the trifluoromethanesulfonate starting materials with respect to thia-Fries rearrangement and free benzyne formation in the same pot.
In conclusion, we have discovered a new reaction of 2-(trimethylsilyl)aryl trifluoromethanesulfonate benzyne precursors, where they bifurcate between thia-Fries rearrangement and aryne generation on simple fluoride treatment. Subsequent union and cyclisation affords novel phenoxathiin-dioxides. This pathway will be relevant to the design of new aryne reaction systems in general, where it may manifest when halogen or nitro-containing aryne precursors are used to explore substrate scope.11 Finally, the reaction affords phenoxathiin-dioxides by a new route that does not require the preparation of a phenoxathiin and subsequent oxidation.
We thank the University of Manchester and the EPSRC for funding (Leadership fellowship to M.F.G.). Dr Anna Collins (University of Edinburgh) is thanked for X-ray crystallography and we thank the EPSRC mass spectrometry service at the University of Swansea.
Notes and references
- (a) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 Search PubMed; (b) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766–3778 Search PubMed; (c) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC; (d) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 RSC.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211–1214 CAS.
- (a) T. Pirali, F. Z. Zhang, A. H. Miller, J. L. Head, D. McAusland and M. F. Greaney, Angew. Chem., Int. Ed., 2012, 51, 1006–1009 Search PubMed; (b) D. McAusland, S. Seo, D. G. Pintori, J. Finlayson and M. F. Greaney, Org. Lett., 2011, 13, 3667–3669 CAS; (c) K. Biswas and M. F. Greaney, Org. Lett., 2011, 13, 4946–4949 CrossRef CAS; (d) A. A. Cant, L. Roberts and M. F. Greaney, Chem. Commun., 2010, 46, 8671–8673 RSC; (e) A. A. Cant, G. H. V. Bertrand, J. L. Henderson, L. Roberts and M. F. Greaney, Angew. Chem., Int. Ed., 2009, 48, 5199–5202 CrossRef CAS.
- CCDC 945230 contains the crystallographic data for 2a. ORTEP-3 was used to produce the thermal ellipsoid plots: L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 Search PubMed.
- For the development of phenoxathiin-dioxides as monoamine oxidase inhibitors, see: M. Harfenist, D. P. C. Mcgee and H. L. White, J. Med. Chem., 1991, 34, 2931–2933 Search PubMed.
- Intramolecular OAr → CAr sulfone transfer has been established in the anionic thia-Fries rearrangement by Lloyd-Jones and co-workers, using a crossover experiment with labelled substrates: A. M. Dyke, D. M. Gill, J. N. Harvey, A. J. Hester, G. C. Lloyd-Jones, M. P. Munoz and I. R. Shepperson, Angew. Chem., Int. Ed., 2008, 47, 5067–5070 Search PubMed.
- J. P. H. Charmant, A. M. Dyke and G. C. Lloyd-Jones, Chem. Commun., 2003, 380–381 RSC.
- (a) Z. Zhao, J. Messinger, U. Schön, W. Rudolf and H. Butenschön, Chem. Commun., 2006, 3007–3009 RSC; (b) G. Werner, C. W. Lehmann and H. Butenschön, Adv. Synth. Catal., 2010, 352, 1345–1355 CrossRef CAS; (c) G. Werner and H. Butenschön, Eur. J. Org. Chem., 2012, 3132–3141 Search PubMed.
- Other examples of the anionic thia-Fries rearrangement: (a) I. R. Hardcastle, PhD thesis, University of Manchester, 1990; (b) R. Kargbo, Y. Takahashi, S. Bhor, G. R. Cook, G. C. Lloyd-Jones and I. R. Shepperson, J. Am. Chem. Soc., 2007, 129, 3846–3847 CrossRef CAS; (c) X. H. Xu, X. Wang, G. K. Liu, E. Tokunaga and N. Shibata, Org. Lett., 2012, 14, 2544–2547 Search PubMed; (d) E. Yoshioka and H. Miyabe, Tetrahedron, 2012, 68, 179–189 Search PubMed; (e) X. H. Xu, M. Taniguchi, A. Azuma, G. K. Liu, E. Tokunaga and N. Shibata, Org. Lett., 2013, 15, 686–689 Search PubMed.
- R. W. Steensma, S. Galabi, J. R. Tagat and S. W. McCombie, Tetrahedron Lett., 2001, 42, 2281–2283 CrossRef CAS.
- For examples of 6-halo-2-trimethylsilyl(phenyl) trifluoromethanesulfonates used with fluoride to generate arynes, see: (a) J. D. Kirkham, P. M. Delaney, G. J. Ellames, E. C. Row and J. P. A. Harrity, Chem. Commun., 2010, 46, 5154–5156 RSC; (b) B. Lakshmi, U. K. Wefelscheid and U. Kazmaier, Synlett, 2011, 345–348 Search PubMed; (c) D. Rodriguez, A. Cobas, D. Peña, D. Perez and E. Guitian, Org. Lett., 2012, 14, 1363–1365 Search PubMed; (d) Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955–2958 Search PubMed.
Footnotes |
| † Celebrating 300 years of Chemistry at Edinburgh. |
| ‡ Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. CCDC 945230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc44529a |
| This journal is © The Royal Society of Chemistry 2013 |