 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-mediated oxidative decarboxylation of arylacetic acids into benzyl radicals: addition to electron-deficient alkenes by using photoredox catalysts†
Yoshihiro
Miyake‡
*,
Kazunari
Nakajima
and
Yoshiaki
Nishibayashi
*
Institute of Engineering Innovation, School of Engineering, The University of Tokyo, Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ynishiba@sogo.t.u-tokyo.ac.jp; Fax: +81 3-5841-1175; Tel: +81 3-5841-1175
First published on 4th July 2013
Abstract
Reactions of arylacetic acids bearing an amino group at the para-position in the benzene ring with alkenes in the presence of a catalytic amount of transition metal polypyridyl complexes as photocatalysts under visible light illumination proceed smoothly to give the corresponding benzylated products via oxidative decarboxylation in good to high yields.
Oxidative decarboxylation of carboxylic acids is one of the fundamental and important processes in nature. The mechanism of decarboxylation has been extensively studied in biochemistry.1 In organic chemistry, decarboxylation under electrochemical and photochemical conditions provides a useful approach to the formation of alkyl radicals, therefore the decarboxylative dimerization has been found to occur via alkyl radicals as reaction intermediates.2,3 The selective bond cleavage at the β-position with respect to the radical cation center triggered by photoinduced electron transfer is expected to be one of the most useful methods for the decarboxylative formation of alkyl radicals from carboxylic acids.4 Especially, it is well known that a single electron oxidation of α-amino acids gives α-aminoalkyl radicals via decarboxylation, and synthetic use of the α-aminoalkyl radicals has been reported (Scheme 1a).5,6 In contrast, synthetic utilization of the aminoarylacetic acids, which are the “phenylogue” of the α-amino acids, has been quite limited6a,7,8 in spite of the utility of the generated benzyl radicals bearing an amino group in organic synthesis (Scheme 1b).9
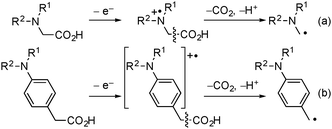 | ||
| Scheme 1 Oxidative decarboxylation of carboxylic acids bearing an amino group. | ||
Recently we have succeeded in the utilization of α-aminoalkyl radicals via fragmentation of radical cations10 by a single electron oxidation of amines mediated by photocatalysts.11–13 We have envisaged that a single electron transfer using photocatalysts is suitable for oxidation of aminoarylacetic acids to trigger decarboxylation resulting in the corresponding benzyl radicals (Scheme 1b). In fact, we have found radical addition reactions via decarboxylation of the aminoarylacetic acids to electron-deficient alkenes. Preliminary results are described herein.
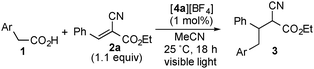
At first, we investigated the reaction of 4-(N,N-dimethylamino)phenylacetic acid (1a) with ethyl (E)-2-cyano-3-phenylpropenoate (2a) under various conditions (Table 1). When a solution of 1a with 1.1 equiv. of 2a in the presence of [4a][BF4] (1 mol%) in acetonitrile (MeCN) was illuminated with a white LED (14 W) at 25 °C for 18 h, ethyl 2-cyano-4-(4-(N,N-dimethylamino)phenyl)-3-phenylbutanoate (3a) was obtained in 85% yield (Table 1, entry 1). The reaction using [4b][BF4] as a photocatalyst also proceeded smoothly to give 3a in a similar yield (Table 1, entry 2), while the yield of 3a slightly decreased with the use of [4c][BF4] or [Ru(bpy)3][BF4]2 as a photocatalyst (Table 1, entries 3 and 4). In sharp contrast, when 9,10-dicyanoanthracene (DCA) was used in place of [4a][BF4], no formation of 3a was observed at all (Table 1, entry 5). Use of dimethyl sulfoxide (DMSO) as a solvent in place of MeCN afforded 3a in 81% yield (Table 1, entry 6). When other polar solvents such as N,N-dimethylformamide (DMF) and methanol were used, 3a was obtained in lower yields (Table 1, entries 7 and 8). Using ethyl 4-(N,N-dimethylamino)phenylacetate (5a) under similar conditions did not afford 3a at all (Table 1, entry 9). Separately, we confirmed that no formation of 3a was observed in the absence of visible light or a photocatalyst. In the previously reported oxidative decarboxylation,2,3,7 the homo-coupling of benzyl radicals easily occurred to give the corresponding bibenzyl (6), while no formation of 6 was observed in all cases presented in Table 1. This result indicates that a visible light-mediated electron transfer using photocatalysts is favorable for addition of benzyl radicals derived from phenylacetic acids to alkenes.
|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Solvent | Yield of 3ab (%) |
| a All reactions of 1a (0.25 mmol) with 2a (0.275 mmol) were carried out in the presence of a photocatalyst (0.0025 mmol) in solvent (2.5 mL) under 14 W white LED illumination at 25 °C for 18 h. b Isolated yield. c 5a was used in place of 1a. | |||
| 1 | [4a][BF4] | MeCN | 85 |
| 2 | [4b][BF4] | MeCN | 84 |
| 3 | [4c][BF4] | MeCN | 70 |
| 4 | [Ru(bpy)3][BF4]2 | MeCN | 68 |
| 5 | DCA | MeCN | 0 |
| 6 | [4a][BF4] | DMSO | 81 |
| 7 | [4a][BF4] | DMF | 76 |
| 8 | [4a][BF4] | MeOH | 57 |
| 9c | [4a][BF4] | MeCN | 0 |
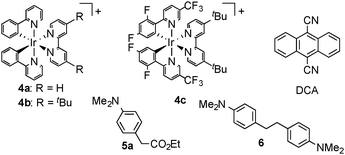
|
|||
Next, we investigated reactions of a variety of arylacetic acids (1) with 2a (Table 2). The reaction of phenylacetic acid bearing an N,N-diethylamino moiety at the para-position of the benzene ring (1b) proceeded to give 3b in 84% yield (Table 2, entry 1). Other phenylacetic acids bearing a variety of tertiary amino groups such as N-methyl-N-phenylamino, N-morpholino, and N-pyrrolidino moieties at the para-position of the benzene ring were also applicable, giving the corresponding products (3c–3e) in high yields (Table 2, entries 2–4). Reactions of phenylacetic acids bearing secondary (1f) and primary amino (1g) groups also took place smoothly to give 3f and 3g in 81% and 87% yields, respectively (Table 2, entries 5 and 6). When the acetic acid bearing a 4-(N-morpholino)-1-naphthyl (1h) group was used, 3h was obtained in a good yield (Table 2, entry 7).
|
|
||
|---|---|---|
| Entry | Arylacetic acid (1) | Yield of 3b (%) |
| a All reactions of 1 (0.25 mmol) with 2a (0.275 mmol) were carried out in the presence of [4a][BF4] (0.0025 mmol) in MeCN (2.5 mL) under 14 W white LED illumination at 25 °C for 18 h. b Isolated yield. c DMSO was used in place of MeCN. | ||
| 1 | Ar = 4-Et2N-C6H4 (1b) | 84 (3b) |
| 2 | Ar = 4-PhMeN-C6H4 (1c) | 89 (3c) |
| 3 | Ar = 4-(N-morpholino)-C6H4 (1d) | 96 (3d) |
| 4 | Ar = 4-(N-pyrrolidino)-C6H4 (1e) | 74 (3e) |
| 5 | Ar = 4-BnHN-C6H4 (1f) | 81 (3f) |
| 6c | Ar = 4-H2N-C6H4 (1g) | 87 (3g) |
| 7 | Ar = 4-(N-morpholino)-1-naphthyl (1h) | 61 (3h) |
Other reactions of 1d or 1g with a variety of alkenes (2) were also examined by using [4a][BF4] as a photocatalyst (Table 3). Introduction of substituents such as methoxy, methyl, phenyl, and chloro at the para-position of the benzene ring of 2a is successful in giving the corresponding products (3i–3l) in good to high yields (Table 3, entries 1–4). Alkenes bearing phenylethyl and iso-propyl groups at the β-position were applied to give 3m and 3n in 80% and 51% yields, respectively (Table 3, entries 5 and 6). The reaction of 1d with benzylidenemalononitrile (2h) proceeded smoothly to give 3o in 70% yield (Table 3, entry 7). However, a lower yield of 3p was obtained when diethyl benzylidenemalonate (2i) was used as a substrate (Table 3, entry 8). The arylacetic acid bearing an amino group (1g) was also transformed efficiently into 3q and 3r in 90% and 51% yields (Table 3, entries 9 and 10).
|
|
|||
|---|---|---|---|
| Entry | Arylacetic acid (1) | Alkene (2) | Yield of 3b (%) |
| a All reactions of 1 (0.25 mmol) with 2a (0.275 mmol) were carried out in the presence of [4a][BF4] (0.0025 mmol) in MeCN (2.5 mL) under 14 W white LED illumination at 25 °C for 18 h. b Isolated yield. c DMSO was used in place of MeCN. | |||
| 1 | 1d | R3 = 4-MeOC6H4, E1 = CN, E2 = CO2Et (2b) | 74 (3i) |
| 2 | 1d | R3 = 4-MeC6H4, E1 = CN, E2 = CO2Et (2c) | 93 (3j) |
| 3 | 1d | R3 = 4-PhC6H4, E1 = CN, E2 = CO2Et (2d) | 66 (3k) |
| 4 | 1d | R3 = 4-ClC6H4, E1 = CN, E2 = CO2Et (2e) | 52 (3l) |
| 5 | 1d | R3 = PhCH2CH2, E1 = CN, E2 = CO2Et (2f) | 80 (3m) |
| 6 | 1d | R3 = iPr, E1 = CN, E2 = CO2Et (2g) | 51 (3n) |
| 7 | 1d | R3 = Ph, E1 = E2 = CN (2h) | 70 (3o) |
| 8 | 1d | R3 = Ph, E1 = E2 = CO2Et (2i) | 16 (3p) |
| 9c | 1g | R3 = 4-MeC6H4, E1 = CN, E2 = CO2Et (2c) | 90 (3q) |
| 10c | 1g | R3 = PhCH2CH2, E1 = CN, E2 = CO2Et (2f) | 51 (3r) |
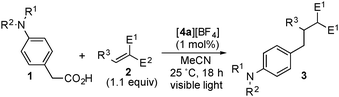
To investigate the effect of the position of an amino group, we carried out reactions of phenylacetic acids bearing a morpholino group at the para-, meta- and ortho-positions of the benzene ring (1d, 1d′ and 1d′′) with 2a under similar conditions (Scheme 2). In sharp contrast to the reaction of 1d, when 1d′ and 1d′′ were used as substrates, no formation of the corresponding products 3d′ and 3d′′ was observed at all. Furthermore, in order to explore the origin of this reactivity, we also investigated fluorescence quenching studies of 4a in the presence of 1d, 1d′ or 1d′′ in MeCN. The rate constants for 1d and 1d′ were estimated to be 2.86 × 108 M−1 s−1 and 6.78 × 107 M−1 s−1 respectively, by using the Stern–Volmer plot, while no quenching of 4a by 1d′′ occurred at all. From these results, it is revealed that oxidation of 1d′ certainly occurred but sequential decarboxylation hardly proceeded. It is reported that the unpaired electron spin density at the para-position in the benzene ring of the one-electron oxidized aniline is much higher than that at the meta-position.14 The radical character in the benzene ring of the one-electron oxidized 1d and 1d′ is expected to have a great effect on the desirable decarboxylation. In the case of 1d′, the radical character of the carbon atom at the meta-position with respect to the amino group (at the β-position with respect to the carboxyl group) of the oxidized 1d′ is too small to induce efficient decarboxylation. On the other hand, Stern–Volmer analysis clearly indicates that oxidation of 1d′′ under photoirradiation occurs scarcely although the spin population at the ortho-position is also known to be similar to that at the para-position. This is probably because an intramolecular hydrogen bonding between the amino group and carboxylic acid in 1d′′ inhibits the electron transfer oxidation.15 In addition, we confirmed that no reaction of 4-methoxyphenylacetic acid (1i) under similar conditions occurred at all. These facts clearly indicate that decarboxylation triggered by a single electron oxidation of the amino group at the para-position is one of the key steps to promote these transformations.
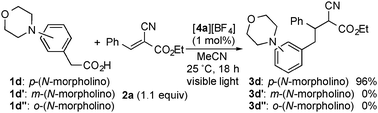 | ||
| Scheme 2 Effect of the position of the amino group on reactions of 1d, 1d′, and 1d′′ with 2a. | ||
A plausible reaction pathway is shown in Scheme 3. First, a single electron oxidation of aminoarylacetic acid (1) by an excited photocatalyst (*cat) and sequential decarboxylation occur to give the benzyl radical (A) and the reduced form of the photocatalyst (cat−).4,8 Addition of A to 2 results in the formation of the radical intermediate (B). Finally, reduction of B by cat− and subsequent protonation proceed to give the product (3) accompanied by regeneration of the photocatalyst (cat). The reduction of B by the reduced form of an iridium catalyst ([Ir(ppy)2(bpy)]0) is feasible according to the literature redox potentials.16,17 The quantum yield in the reaction of 1a with 2a was estimated to be 0.21, which is less than 1 and is in the common range of molecular transformations via photoinduced electron transfer with transition metal polypyridyl complexes.10 This result suggests that the contribution of a photo independent process including a radical chain process is negligible in the present system.10 The pathway shown in Scheme 3 is similar to that described in our previous report on the addition of α-aminoalkyl radicals to electron-deficient alkenes.10c On the other hand, considering the redox potential of cyanoesters 2,16 the reaction pathway via reduction of 2 by the excited photocatalyst17 and subsequent radical–radical coupling is also possible.10a,18
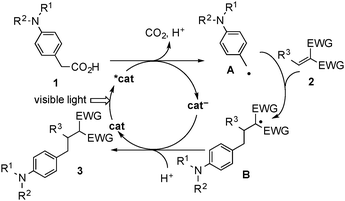 | ||
| Scheme 3 Plausible reaction pathway. | ||
In summary, we have succeeded in the visible light-mediated synthetic utilization of benzyl radicals formed by oxidation and subsequent decarboxylation of arylacetic acids. This reaction system is applicable for addition of a variety of benzyl radicals bearing an amino group at the para-position in the benzene ring to electron-deficient alkenes. A number of transformations using alkyl radicals from alkyl halides as reaction intermediates have been reported until now, where alkyl halides are known to be promising precursors for generation of alkyl radicals.9 However, examples for preparation of alkyl halides bearing amino groups in a single molecule are limited due to undesirable reactions including N-alkylation reactions.19 We believe that the method described here provides an alternative route for generation of benzyl radicals bearing an amino group.
Notes and references
- (a) T. Li, L. Huo, C. Pulley and A. Liu, Bioorg. Chem., 2012, 43, 2 CrossRef CAS; (b) G.-G. Chang and L. Tong, Biochemistry, 2003, 42, 12721 CrossRef CAS; (c) W. W. Cleland, Acc. Chem. Res., 1999, 32, 862 CrossRef CAS.
- A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623 CrossRef CAS.
- D. Budac and P. Wan, J. Photochem. Photobiol., A, 1992, 67, 135 CrossRef CAS.
- (a) E. Baciocchi, M. Bietti and O. Lanzalunga, J. Phys. Org. Chem., 2006, 19, 467 CrossRef CAS; (b) E. Baciocchi, M. Bietti and O. Lanzalunga, Acc. Chem. Res., 2000, 33, 243 CrossRef CAS.
- For reviews, see: (a) D. W. Cho, U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 2011, 44, 204 CrossRef CAS; (b) U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 1992, 25, 233 CrossRef CAS.
- (a) Y. Yoshimi, K. Kobayashi, H. Kamakura, K. Nishikawa, Y. Haga, K. Maeda, T. Morita, T. Itou, Y. Okada and M. Hatanaka, Tetrahedron Lett., 2010, 51, 2332 CrossRef CAS; (b) A. G. Griesbeck, T. Heinrich, M. Oelgemöller, J. Lex and A. Molis, J. Am. Chem. Soc., 2002, 124, 10972 CrossRef CAS; (c) A. G. Griesbeck, T. Heinrich, M. Oelgemöller, A. Molis and A. Heidtmann, Helv. Chim. Acta, 2002, 85, 4561 CrossRef CAS; (d) Z. Su, P. S. Mariano, D. E. Falvey, U. C. Yoon and S. W. Oh, J. Am. Chem. Soc., 1998, 120, 10676 CrossRef CAS.
- L. Cermenati, M. Mella and A. Albini, Tetrahedron, 1998, 54, 2575 CrossRef CAS.
- M. A. Smitha, E. Prasad and K. R. Gopidas, J. Am. Chem. Soc., 2001, 123, 1159 CrossRef CAS.
- (a) For reviews see: P. Renaud, in Radicals in Organic Synthesis, ed. P. Renaud, M. P. Sibi, Wiley-VCH, Weinheim, 2001 Search PubMed; (b) G. J. Rowlands, Tetrahedron, 2010, 66, 1593 CrossRef CAS; (c) I. Ryu, N. Sonoda and D. P. Curran, Chem. Rev., 1996, 96, 177 CrossRef CAS; (d) C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237 CrossRef CAS.
- (a) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem.–Eur. J., 2012, 18, 16473 CrossRef CAS; (b) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966 RSC; (c) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS.
- (a) L. R. Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107 CrossRef; (b) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS; (c) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS; (d) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS.
- For recent reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322, DOI:10.1021/cr300503r; (b) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS; (d) M. A. Ischay and T. P. Yoon, Eur. J. Org. Chem., 2012, 3359 CrossRef CAS; (e) S. Maity and N. Zheng, Synlett, 2012, 1851 CAS; (f) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS.
- G. D'Aprano, E. Proynov, M. Lebœuf, M. Leclerc and D. R. Salahub, J. Am. Chem. Soc., 1996, 118, 9736 CrossRef CAS.
- (a) D. A. Smith and S. Vijayakumar, Tetrahedron Lett., 1991, 32, 3617 CrossRef CAS; (b) X. Li, Y.-D. Wu and D. Yang, Acc. Chem. Res., 2008, 41, 1428 CrossRef CAS; (c) X.-J. Liao, W. Guo and S.-H. Xu, Acta Crystallogr., Sect. E, 2011, 67, o1732 CAS; (d) R. K. Castellano, Y. Li, E. A. Homan, A. J. Lampkins, I. V. Marín and K. A. Abboud, Eur. J. Org. Chem., 2012, 4483 CrossRef CAS.
- X.-Q. Zhu, M. Zhang, Q.-Y. Liu, X.-X. Wang, J.-Y. Zhang and J.-P. Cheng, Angew. Chem., Int. Ed., 2006, 45, 3954 CrossRef CAS.
- F. O. Garces, K. A. King and R. J. Watts, Inorg. Chem., 1988, 27, 3464 CrossRef CAS.
- See ESI† for experimental details.
- Examples for preparation of 4-aminobenzyl bromides: (a) G. Wang, X. Zhang, J. Geng, K. Li, D. Ding, K.-Y. Pu, L. Cai, Y.-H. Lai and B. Liu, Chem.–Eur. J., 2012, 18, 9705 CrossRef CAS; (b) J.-P. Leclerc, J.-P. Falgueyret, M. Girardin, J. Guay, S. Guiral, Z. Huang, C. S. Li, R. Oballa, Y. K. Ramtohul, K. Skorey, P. Tawa, H. Wang and L. Zhang, Bioorg. Med. Chem. Lett., 2011, 21, 6505 CrossRef CAS; (c) H. Enomoto, Y. Morikawa, Y. Miyake, F. Tsuji, M. Mizuchi, H. Suhara, K. Fujimura, M. Horiuchi and M. Ban, Bioorg. Med. Chem. Lett., 2009, 19, 442 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all new compounds. See DOI: 10.1039/c3cc44438d |
| ‡ Current address: Department of Applied Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan. E-mail: miyake@apchem.nagoya-u.ac.jp |
| This journal is © The Royal Society of Chemistry 2013 |