 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reusable and highly active supported copper(I)–NHC catalysts for Click chemistry†
John-Michael Collinson,
James D. E. T. Wilton-Ely* and
Silvia Díez-González*
Department of Chemistry, Imperial College London, South Kensington Campus, London SW7 2AZ, UK. E-mail: j.wilton-ely@imperial.ac.uk; s.diez-gonzalez@imperial.ac.uk; Fax: +44 (0)20 7594 5804; Tel: +44 (0)20 7594 9718 Tel: +44 (0)20 7594 9699
First published on 28th June 2013
Abstract
Immobilised [Cu(NHC)] catalysts are reported for the preparation of 1,2,3-triazoles. In addition to showing outstanding catalytic activity, the catalyst systems are easy to prepare and can be recycled many times.
The copper-mediated cycloaddition reaction of azides and alkynes is a recent example of how metal catalysis can have an impact on a wide range of scientific disciplines.1 It has been thrust into the spotlight by its use as a proof of concept for the Click philosophy postulated by Sharpless.2 The reported applications for this cycloaddition reaction are numerous and diverse, but the development of true Click transformations still relies on the significant efforts being devoted to improve the understanding of the mechanism(s) involved,3 as well as on the development of ligand-modified systems.4 Among the families of ligands reported for this reaction, N-heterocyclic carbenes (NHCs)5 are prominent as they produce highly robust catalysts capable of conferring outstanding catalytic activity on the copper centre, often at extremely low catalytic loadings.6
The immobilisation of metals on nanoparticles allows the properties and flexibility of a metal–ligand unit to be harnessed while exploiting the advantages of attachment to a surface.7 An enhanced Click catalyst for the cycloaddition of azides and alkynes could thus be achieved by a system that combines the exceptional activity of copper–NHC systems with the easy separation and recyclability of a heterogeneous catalyst.8 In this quest, it was decided to use [CuI(IAd)] (IAd = 1,3-di(adamantyl)imidazol-2-ylidene) as the starting point due to its superior reactivity when compared to other neutral [Cu(NHC)] complexes.6d Thus, it was anticipated that an analogue of the IAd ligand could be immobilised on silica through a hydroxyl substituent on the NHC backbone. Precedents in the literature for such architectures (sometimes referred to as switchable carbenes) include copper(I) complexes.9 More importantly, the modification of the ligand backbone minimises the changes around the metal centre, as significantly diminished catalytic activities are often reported for catalysts attached to surfaces via the substituent on the N atoms of the NHC ligand.10
Following the reported synthetic route for the preparation of 4-hydroxyimidazol-2-ium salts bearing aromatic groups on the nitrogen atoms,9c the known adamantylformamidine 1 was prepared.11 However, under acylation conditions 1 decomposed to give 2 as the sole isolated product (Scheme 1).12 This reactivity might be attributed to the fragility of alkyl formamidines when compared to their aryl analogues.13 Thus, construction of the ligand was commenced instead from the backbone. The preparation of the ligand OHIAd·HBF4 5·HBF4 was then accomplished in three simple steps from adamantyl amine, starting with its acylation by chloroacetyl chloride, followed by amine alkylation (Scheme 1). Subsequent cyclisation with triethyl orthoformate led to the isolation of 5·HBF4. Gratifyingly, it was found that all these reactions could be run in air with no need for purification on silica gel.
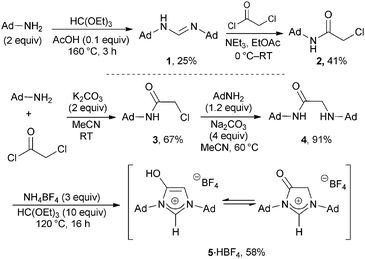 | ||
| Scheme 1 Synthesis of OHIAd·HBF4, 5·HBF4. | ||
It is important to note that, for known 4-hydroxyimidazolium ligands (and their 4-amino analogues),14 the enol tautomer is so favourable that no 4-oxo-1,3-diarylimidazolium derivatives are observed. In contrast, the 1H NMR spectrum of 5·HBF4 showed two singlets at 6.16 and 4.10 ppm, attributed to the enol and keto tautomers, respectively. The integration of these signals indicated a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equilibrium between both forms of OHIAd·HBF4.
:1 equilibrium between both forms of OHIAd·HBF4.
The new azolium salt was then immobilised on three different carrier materials, namely silica flakes, silica nanoparticles and magnetite/silica nanoparticles. This was achieved by reaction of these supports with 5·HBF4 in hot DMF in the presence of molecular sieves. The anchored ligands were then treated with CuI under otherwise identical conditions to those reported for the preparation of [CuI(IAd)] to give the corresponding supported [CuI(5)] complexes (Scheme 2).
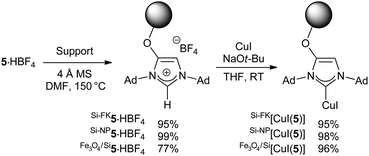 | ||
| Scheme 2 Preparation of supported catalysts. Mass balances are reported. | ||
Transmission Electron Microscopy (TEM) revealed that the silica nanoparticles, Si-NP[CuI(5)], possessed an average diameter of 150.4 (±5.3) nm, while those with a magnetite core, Fe3O4/Si[CuI(5)], were 47.7 (±3.8) nm in size. All three functionalised silica materials showed the presence of copper, as determined by Energy Dispersive X-Ray Spectroscopy (EDS). Infrared spectroscopy provided limited information due to the small proportion of surface units compared to the bulk material of the nanoparticles. Initially, mass balance calculations were used to determine the loading of copper surface units on the nanoparticles but this was replaced by Inductively Coupled Plasma (ICP) analysis to provide the mmolCu per mg loading for the materials.
With the supported copper complexes in hand, the influence of the support on the catalytic activity was examined, along with the recycling efficiency of each system. Hence, the three prepared catalysts were applied to the model reaction of benzyl azide 6a and phenylacetylene on water at room temperature (Scheme 3). In the first cycle, all catalysts led to the quantitative formation of triazole 7a after overnight reactions with only 1 mol% [Cu]. After dissolving the formed triazoles in EtOAc, the reaction mixtures were centrifuged, and the catalysts were collected by filtration or using a magnet, washed, dried under vacuum and reused in a new cycle. The catalyst supported on silica flakes lost its catalytic activity by the fifth cycle, whereas the copper-functionalised silica nanoparticles led to diminished conversions after only 3 cycles (53 and 68% conversion, respectively). However, the copper complex anchored on coated magnetic nanoparticles displayed optimal activity for at least nine cycles before deactivation (91, 82 and 20% conversions for cycles 9 to 11). The improved recyclability observed for Fe3O4/Si[CuI(5)] is attributed to its facile manipulation and recovery by simple decantation in the presence of a hand-held magnet.
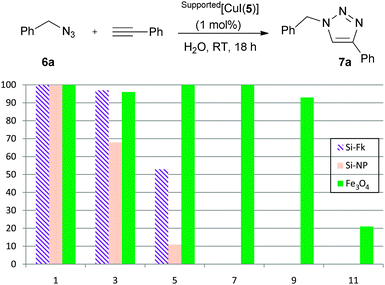 | ||
| Scheme 3 Recyclability of supported catalysts. 1H NMR conversions are the average of at least two independent experiments. | ||
To highlight the essential role of the NHC ligand, silica flakes were treated with CuI in the absence of the NHC ligand, resulting in a yield for 7a of only 56% under otherwise identical reaction conditions. This result clearly showed that the observed catalytic activity can be attributed to supported NHC–copper species.
Optimisation studies revealed that Fe3O4/Si[CuI(5)] performed equally well in the model reaction with only 0.25 mol% copper loading and this finding was used in the further investigation of the scope of the reaction (Table 1). When incomplete conversions were observed, either a slightly higher copper loading (0.5 mol%) or a small increase in the reaction temperature (40 °C) allowed the expected triazoles to be formed quantitatively.
|
|||
|---|---|---|---|
| Entry | Triazole | [Cu]/mol%, T/°C | Yielda/% |
| a Isolated yields are the average of at least two independent experiments.b 1H NMR conversions are the average of at least two independent experiments. | |||
| 1 |  |
0.25, RT | 92 |
| 2 | 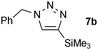 |
0.25, RT | 40b |
| 0.5, RT | 80 | ||
| 0.25, 40 °C | 83 | ||
| 3 | 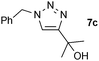 |
0.25, RT | 70b |
| 0.25, 40 °C | 89 | ||
| 4 | 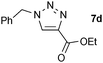 |
0.25, RT | 74b |
| 0.25, 40 °C | 97 | ||
| 5 | 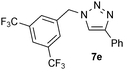 |
0.25, RT | 26b |
| 0.25, 40 °C | 89 | ||
| 6 | 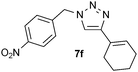 |
0.25, RT | 5b |
| 0.25, 40 °C | 65 | ||
| 7 | 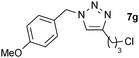 |
0.25, RT | 26b |
| 0.5, RT | 96 | ||
| 0.25, 40 °C | 81 | ||
| 8 | 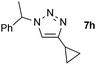 |
0.25, RT | 72b |
| 0.25, 40 °C | 84 | ||
| 9 | 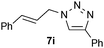 |
0.25, RT | 85b |
| 0.25, 40 °C | 89 | ||
| 10 | 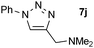 |
0.25, RT | 87 |
| 11 | 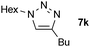 |
0.25, RT | 93 |
| 12 | 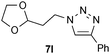 |
0.25, RT | 35b |
| 0.25, 40 °C | 88 | ||
| 13 | 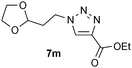 |
0.25, RT | 82b |
| 0.25, 40 °C | 98 | ||
| 14 | 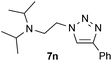 |
0.25, RT | 87 |
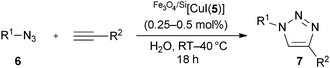
Overall, a variety of triazoles 7 was isolated in excellent yields and high purity after simple extraction and decantation/filtration, regardless of the substitution on either the starting azide 6 or alkyne. Several functional groups (e.g., alcohol, amine, halogen) were well tolerated by benzylic, aromatic or aliphatic azides. Increasing the steric profile of either of the cycloaddition partners did not diminish the catalytic activity (Table 1, entries 3 and 8).
The performance of Fe3O4/Si[CuI(5)] was also compared to the parent [CuI(IAd)], probably one of the best reported catalysts for this particular cycloaddition. Both complexes led to complete conversion to 7a (model reaction, see Scheme 3) with only 0.25 mol% [Cu] at room temperature. In order to probe the relative activity further, another pair of cycloaddition partners was chosen that did not lead to complete conversion with the supported catalyst under such reaction conditions (Scheme 4, see also Table 1, entry 4). Using either [CuI(IAd)] or Fe3O4/Si[CuI(5)] produced 7d at a comparable level of conversion, suggesting that the catalytic activity of Fe3O4/Si[CuI(5)] is not affected significantly by its immobilised nature.
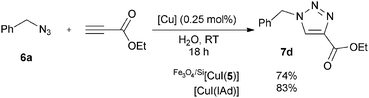 | ||
| Scheme 4 Comparison of copper catalysts. | ||
ICP analysis of 7h formed using [CuI(IAd)] showed that it still contained all the starting copper loading, even after acid hydrolysis, whereas the same analysis of 7h produced using Fe3O4/Si[CuI(5)] showed extremely low copper contamination (around 3% of the original metal loading). TEM and EDS analysis of the used Cu–silica–magnetite catalyst demonstrated no discernible change of structure and showed no evidence of formation of copper nanoparticles.
In conclusion, a supported copper(I) catalyst has been developed that combines high recyclability with the remarkable activity of [Cu(NHC)] systems in the cycloaddition reaction of azides and alkynes. Importantly, in terms of its wider utility, the new system does not require a lengthy or complicated synthetic sequence for its preparation. The activity of the supported catalyst is comparable to its homogeneous analogue while permitting the reuse of the catalyst up to ten times after simple decantation in the presence of a hand-held magnet.
Imperial College and EPSRC are gratefully acknowledged for financial support of this work.
Notes and references
- For seminal reports, see: (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS . For the first report on a copper(I)-catalyzed [3+2] cycloaddition reaction, see: ; (c) G. L'abbe, Bull. Soc. Chim. Belg., 1984, 93, 579–592 CrossRef CAS . Special issue on Click chemistry, see: ; (d) Chem. Soc. Rev., 2010, 39, 1221–1408. For reviews on diverse applications, see: ; (e) W. H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675–696 RSC; (f) M. Glaser and E. G. Robins, J. Labelled Compd. Radiopharm., 2009, 52, 407–414 CrossRef CAS; (g) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS; (h) G. Franc and A. Kakkar, Chem. Commun., 2008, 5267–5276 RSC; (i) J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS; (j) A. Dondoni, Chem.–Asian J., 2007, 2, 700–708 CrossRef CAS; (k) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- For a very recent example, see: (a) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS . For a review, see: ; (b) V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2005, 51–68 Search PubMed.
- S. Díez-González, Catal. Sci. Technol., 2011, 1, 166–178 Search PubMed.
- (a) N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. P. Díez-González, RSC Catalysis Series, Cambridge, 2011 Search PubMed; (b) Special issue on carbenes, Chem Rev., 2009, 109, 3209–3884; (c) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem.–Eur. J., 2006, 12, 7558–7564 CrossRef; (b) S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2008, 47, 8881–8884 CrossRef; (c) M. L. Teyssot, A. Chevry, M. Traikia, M. El-Ghozzi, D. Avignant and A. Gautier, Chem.–Eur. J., 2009, 15, 6322–6326 CrossRef CAS; (d) S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC; (e) T. Nakamura, T. Terashima, K. Ogata and S.-i. Fukuzawa, Org. Lett., 2011, 13, 620–623 CrossRef CAS; (f) F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969–7975 CrossRef CAS.
- (a) E. R. Knight, N. H. Leung, A. L. Thompson, G. Hogarth and J. D. E. T. Wilton-Ely, Inorg. Chem., 2009, 48, 3866–3874 CrossRef CAS; (b) S. Naeem, A. Ribes, A. J. P. White, M. N. Haque, K. B. Holt and J. D. E. T. Wilton-Ely, Inorg. Chem., 2013, 52, 4700–4713 CrossRef CAS; (c) J. D. E. T. Wilton-Ely, Dalton Trans., 2008, 25–29 RSC; (d) E. K. Beloglazkina, A. G. Majouga, R. B. Romashkina, N. V. Zyk and N. S Zefirov, Russ. Chem. Rev., 2012, 81, 65–90 CrossRef CAS; (e) S. Roy and M. A. Pericàs, Org. Biomol. Chem., 2009, 7, 2669–2677 RSC.
- For a critical review, see: B. Dervaux and F. E. Du Prez, Chem. Sci., 2012, 3, 959–966 RSC . For previous approaches, see: (a) P. Li, L. Wang and Y. Zhang, Tetrahedron, 2008, 64, 10825–10830 CrossRef CAS; (b) G. M. Pawar, B. Bantu, J. Weckesser, S. Blechert, K. Wurst and M. R. Buchmeiser, Dalton Trans., 2009, 9043–9051 RSC; (c) W. Wang, J. Wu, C. Xia and F. Li, Green Chem., 2011, 13, 3440–3445 RSC.
- (a) L. Benhamou, V. César, H. Gornitzka, N. Lugan and G. Lavigne, Chem. Commun., 2009, 4720–4722 RSC; (b) A. T. Biju, K. Hirano, R. Frçhlich and F. Glorius, Chem.–Asian J., 2009, 4, 1786–1789 CAS; (c) L. Benhamou, N. Vujkovic, V. César, H. Gornitzka, N. Lugan and G. Lavigne, Organometallics, 2010, 29, 2616–2630 CrossRef CAS; (d) A. S. K. Hashmi, C. Lothschütz, K. Graf, T. Häffner, A. Schuster and F. Rominger, Adv. Synth. Catal., 2011, 353, 1407–1412 CrossRef CAS.
- W. J. Sommer and M. Weck, Coord. Chem. Rev., 2007, 251, 860–873 CrossRef CAS.
- M. Braun, W. Frank and C. Ganter, Organometallics, 2012, 31, 1927–1934 CrossRef CAS.
- A. Paczal, A. C. Bényei and A. Kotschy, J. Org. Chem., 2006, 71, 5969–5979 CrossRef CAS.
- E. C. Taylor and W. A. Ehrhart, J. Org. Chem., 1963, 28, 1108–1112 CrossRef CAS.
- V. César, J.-C. Tourneux, N. Vujkovic, R. Brousses, N. Lugan and G. Lavigne, Chem. Commun., 2012, 48, 2349–2351 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and information on characterisation of the materials described. See DOI: 10.1039/c3cc44371j |
| This journal is © The Royal Society of Chemistry 2013 |