 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photovoltaic efficiency via light-triggered self-assembly†
Rohan J.
Kumar
*ab,
Quentin I.
Churches
b,
Jegadesan
Subbiah
ab,
Akhil
Gupta
b,
Abdelsalem
Ali
b,
Richard A.
Evans
b and
Andrew B.
Holmes
ab
aSchool of Chemistry, Bio21 Institute, University of Melbourne, Melbourne, VIC 3010, Australia. E-mail: jkumar@unimelb.edu.au
bCSIRO Materials Science and Engineering, Private Bag 10, Clayton South, Victoria 3169, Australia
First published on 30th May 2013
Abstract
Light-initiated, radical and hydrogen-bond induced self-assembly of bis-acetamido-functionalized triarylamines is demonstrated to occur in strongly dipolar “push–pull” molecules. This self-assembly process results in the formation of self-assembled nanostructures which in turn increase the efficiency of organic photovoltaic devices.
Molecules that respond to external stimuli, triggering functional changes at the macroscopic level, will lead to a new generation of functional materials.1 Organic chemistry offers a broad technological platform2 through which signal response leading to hierarchical organization and ultimately the functional output can be designed and encoded at the molecular scale.3 In fact, organic compounds form the basis of complex and far from equilibrium systems found in nature.
Organic photovoltaics (OPVs) is an emerging technology that seeks to lower the manufacturing costs of photovoltaic devices through rapid solution-based manufacturing techniques.4 This technology is presently limited by the fact that the optimal molecular orientation of active layer components may not be directly accessible,5 introducing the need for additives and post-processing techniques (e.g. thermal6 and solvent7 annealing). Additionally, the molecular orientation in the active layer may change over time, leading to reduced performance. The development of optically responsive molecules for organic photovoltaics that achieve the molecular orientation for optimal performance in response to an external optical stimulus may simultaneously overcome both these problems. If the external stimulus is broad spectrum light, this will be encountered as part of the operating environment of the device.
Molecules for OPVs commonly incorporate an intramolecular dipole to enhance the absorption of light.8 The dipole is introduced by the combination of an electron poor “acceptor” motif and an electron rich “donor” motif. Triarylamines have been widely employed as the donor motif in molecules both for bulk-heterojunctions,8a,c and dye-sensitized solar cells.8b An N-acetamidotriarylamine derivative capable of participating in intermolecular hydrogen bonding interactions has been shown to participate in light-initiated supramolecular assembly9 to form nanostructures that were subsequently incorporated into highly conductive, functional electronic devices.10
Here these approaches are combined in the design of a light-responsive molecule for use in OPVs, incorporating hydrogen-bonding groups into the triarylamine donor motif within a strongly dipolar charge transfer material. It was hypothesized that light-triggered radical cation formation would enable supramolecular association of the hole-transporting triarylamine domains of a p-type organic semiconductor and the consequent superior morphology for photovoltaic devices. The target p-type molecules 4 and 5 contained acetamidotriarylamine groups connected through conjugated linkers to the electron withdrawing cyanopyridone8a (4) and dicyanovinyl (5) substituents. These were prepared (Scheme 1) by conventional Suzuki coupling procedures and Knoevenagel chain extension. The two final compounds varied significantly in their solubility, with compound 4 being soluble in a wide range of organic solvents and compound 5 being only slightly soluble (∼5 mg mL−1) in chlorinated solvents, e.g. chloroform and chlorobenzene. Compound 5 was highly soluble in binary solvent mixtures consisting of alcohols and chlorinated solvents, presumably owing to disruption of the strong hydrogen bonding interactions.
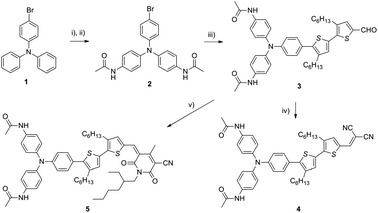 | ||
| Scheme 1 Synthesis. Reagents and conditions: (i) Cu(NO3)2·2.5H2O, Ac2O, 50 °C, 4 h; (ii) SnCl2, Ac2O, 60 °C, 4 h; 62% over 2 steps; (iii) 3,4′-dihexyl-5′-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,2′-bithiophene-5-carbaldehyde, Pd(PPh3)4, Cs2CO3, toluene, reflux, 16 h; 52%; (iv) malononitrile, pyridine, chloroform, reflux, 4 h; 67%; (v) 1-(2-ethylhexyl)-4-methyl-2,6-dioxo-1,2,5,6-tetrahydropyridine-3-carbonitrile, CH3OH, reflux, 2 h; 85%. | ||
The optoelectronic and electrochemical properties of compounds 4 and 5 were investigated in freshly prepared chloroform solution and as neat films prepared in the dark (see ESI,† Fig. S1). The frontier orbital energy levels were determined via cyclic voltammetry or alternatively photoelectron spectroscopy in air (PESA) and the energy was estimated from the absorption onset of the UV/Vis absorption spectrum (Table 1). For both compounds 4 and 5, the calculated energy levels are favourable for incorporation into OPV devices with PC61BM as the electron acceptor.11
| Compound | Absorption (nm) | Emissiona (nm) |
E
½![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b(ΔEp)c mV b(ΔEp)c mV |
HOMOd (eV) |
E
ge (eV) |
LUMOf (eV) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ max (CHCl3) | λ max (film) | λ max (CHCl3) | λ max (film) | R/R− | R+/R | R2+/R | ||||
| a Emission recorded using excitation at absorption maxima. b Half wave potential determined as the average of the anodic and cathodic peak potentials, V vs. ferrocene 10 mM in CH2Cl2, 0.1 M Bu4N(PF6). c Difference between the anodic and cathodic peak potentials, V, v = 0.1 mV s−1. d Determined from EHOMO = −(Eox + 4.80) eV, data in brackets measured by PESA on thin films. e Determined from the difference in the electrochemically measured HOMO and LUMO; the value in brackets estimated from the onset of absorption in thin films. f Determined from ELUMO = −(Ered + 4.80) eV, data in brackets measured by PESA on thin films and absorption onset ELUMO = EHOMO + Eg. g Irreversible reduction, the value of the peak reduction potential. | ||||||||||
| 4 | 499, 326 | 513 | 701 | 720 | −1.6g | 0.30 (0.18) | — | −5.1 | 1.9 (1.9) | −3.2 |
| 5 | 571, 323 | 584 | 754 | 786 | −1.2g | 0.27 (0.058) | 0.73 (0.090) | −5.1 (−5.4) | 1.5 (1.75) | −3.6 (−3.7) |
The self-assembly behaviour of compounds 4 and 5 was then investigated via1H-NMR studies in CDCl3. The 1H-NMR spectrum of a freshly prepared sample (5 mM) shows the expected peaks and relative intensities; however, upon irradiation of the sample under white light for two hours, the resonances of the aromatic ring protons on and proximal to the triarylamine motif broaden and diminish in relative intensity with respect to those of the freshly prepared sample (Fig. 1). The change in 1H-NMR spectra was shown to be thermally reversible and takes place in degassed solutions (see ESI,† Fig. S2), in accord with the original hypothesis of Giuseppone et al.9 Control solutions of 4 and 5 in CDCl3 that were not subject to irradiation over the same time period did not show any change in the 1H-NMR spectrum. This behaviour is assigned to the self-assembly of the molecules due to the combined effects of hydrogen bonding and a charge transfer interaction of the photo-generated radical cation with its neutral counterpart, as initially described by Giuseppone et al.9,10 This behaviour was replicated in CHCl3 solution in the presence of PC61BM (see ESI,† Fig. S3), showing that this self-assembly pathway is not disrupted by the presence of PC61BM.
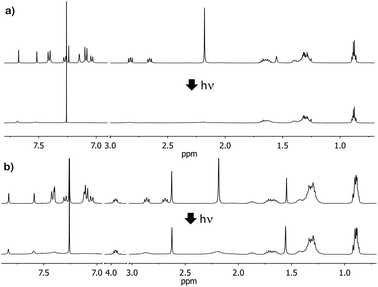 | ||
| Fig. 1 Change in the 1H-NMR spectrum of a 5 mM solution of 4 (a) and 5 (b) in CDCl3 following 2 hours of irradiation with white light (tungsten source). | ||
Following the 1H-NMR experiments, the EPR and UV/Vis absorption and emission spectra of solutions of the irradiated samples were obtained. EPR analysis confirmed the presence of triarylamine radicals in irradiated solutions only for both compounds 4 and 5. Only minor differences were observed in the UV/Vis spectra between irradiated and freshly prepared samples (see ESI,† Fig. S3) suggesting that the photo-induced self-assembly process does not greatly perturb the main electronic transitions of the chromophore.
Visualization of the self-assembled nanostructures of compounds was achieved by atomic force microscopy (AFM, Fig. 2). A solution was prepared and divided into the irradiated and dark samples; the irradiated sample was prepared under white light for 2 hours and the dark sample was kept in the dark during this time. The 1H-NMR spectra of the samples were then checked for self-assembly, evidence for which was observed only for the irradiated sample. The samples were then drop-cast onto silicon wafers and examined by AFM. The structures differed dramatically. For compound 5 samples that were not exposed to light formed semi-circular or donut-shaped aggregates of 200–300 nm in size (Fig. 2a), whereas following irradiation, a change in the morphology of the nanostructures was evident with longer interconnected fibres being present (Fig. 2b).
 | ||
| Fig. 2 Tapping mode AFM images of drop-cast films deposited on silicon from 1 mg mL−1 CHCl3 solutions of 4 and 5. Solutions of 4 were processed (a) under dark conditions and (b) after irradiation with white light (2 h). Solutions of 5 were processed (c) under dark conditions and (d) after irradiation with white light (tungsten source, 2 h). | ||
For compound 4 AFM investigation of drop-cast solution showed no nanostructures before irradiation (Fig. 2c), with only amorphous aggregates observed on the surface. Under irradiated conditions (Fig. 2d), there is some evidence for small nanowires in the order of several nanometers in size, at the limit of the instrument's resolution.
Giuseppone and co-workers reported9 that the initial light-induced stacking of the triarylamine molecules was followed by a second “bundling” step to give nanostructures as observed by AFM in the range of 10–50 nm. Here for compound 5 large structures were observed by AFM under both conditions, indicating that additional, hierarchical self-association takes place, even in the absence of light induced stacking interactions. However, for compound 4, the absence of higher order structures as observed by AFM indicates that the molecules do not spontaneously self-assemble in the absence of light, and that the nanostructures formed in the presence of light do not engage in further self-assembly steps to give higher order species. The difference in the behaviour of the two compounds is possibly due to their different solubility profiles and the different hydrogen bonding properties of cyanopyridone in comparison with the dicyanovinyl acceptor unit. However, films spin-coated from a blended chloroform solution of 4 with PC61BM also show morphological differences (see ESI,† Fig. S6), indicating that the self-assembly observed by 1H-NMR has morphological consequences in films similar to those used in OPV devices.
The functional effect of the nanostructures observed in solution by NMR and AFM was studied in OPV devices. Owing to its low solubility compound 5 proved unsuitable; however, compound 4 was sufficiently soluble to enable devices to be made. Irradiated and dark samples were prepared as previously described and they were mixed in a 1:1 ratio (w/w) with PC61BM in chloroform solution. The presence of self-assembled structures was verified in solution by 1H-NMR spectroscopy before film deposition. This was carried out identically for the two samples thus ensuring that the only difference in the devices was the exposure of the active layer solution to light before deposition. Films were formed by spin-coating the active layer onto a film of nanoparticulate ZnO. The devices were of the architecture glass/ITO/ZnO/active layer/MoO3/Ag and the unoptimized device data are summarized in Table 2.
| Condition | J SC (mA cm−1) | V OC (V) | FFd (%) | Δ (%) |
|---|---|---|---|---|
|
a Data are the mean and standard deviation recorded for 10 devices. Characteristics of devices made from the 1:1 (w/w) ratio of 4 (7.8 mM) and PCBM. The active layer was deposited by spin coating from chloroform.
b Short circuit current.
c Open circuit voltage.
d Fill factor.
e Device photochemical power conversion efficiency (PCE).
|
||||
| Dark | 2.75 ± 0.13 | 0.62 ± 0.01 | 38 ± 3 | 0.65 ± 0.09 |
| Irradiated | 3.10 ± 0.10 | 0.62 ± 0.01 | 46 ± 2 | 0.90 ± 0.07 |
The device data show that the VOC measured from irradiated compared with non-irradiated samples is identical. The overall efficiency of the device fabricated from the irradiated solution is significantly (p = 0.001) higher than that fabricated from a solution not exposed to light. This is due to significant increases in both Jsc (p = 0.001) and fill factor (p = 0.001), parameters that are sensitive to morphologically determined phenomena such as charge transport, collection and recombination. The observation of morphological differences in the AFM experiments leads to the conclusion that the light-initiated self-assembly process leads to the formation of supramolecular networks for charge transport. The application of this phenomenon to OPV devices opens the way for further investigation of, in particular, whether this behaviour is replicable in the presence of an electron acceptor in the solid state, enabling light induced annealing of solar cells.
A diacetamido-triarylamine motif was incorporated into a strongly dipolar “push–pull” small molecule with suitable optoelectronic properties to act as a p-type material in OPV devices. Molecules containing this motif were shown to undergo light initiated self-assembly in chloroform solution, and active layers deposited from solutions of 5 that showed self-assembly afforded higher efficiency OPV devices.
The authors thank the Commonwealth Scientific and Industrial Research Organization, the Australian Solar Institute (ARENA) and the Victorian State Government (DBI-VSA and DPI-ETIS) for generous financial support of the Victorian Organic Solar Cell Consortium. We thank Dr M. Groarke and Mr T. Wilson for their technical assistance and Professor K Ghiggino for helpful discussions.
Notes and references
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed
.
-
(a) P. Charbonnaz, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1492–1496 RSC
-
(a) N. Giuseppone, Acc. Chem. Res., 2012, 45, 2178–2188 CrossRef CAS PubMed
- N. Espinosa, M. Hösel, D. Angmo and F. C. Krebs, Energy Environ. Sci., 2012, 5, 5117–5132 RSC
- M. Grätzel, R. A. J. Janssen, D. B. Mitzi and E. H. Sargent, Nature, 2012, 488, 304–312 CrossRef PubMed
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed
-
(a) A. Gupta, A. Ali, A. Bilic, M. Gao, K. Hegedus, B. Singh, S. E. Watkins, G. J. Wilson, U. Bach and R. A. Evans, Chem. Commun., 2012, 48, 1889–1891 RSC
- E. Moulin, F. Niess, M. Maaloum, E. Buhler, I. Nyrkova and N. Giuseppone, Angew. Chem., Int. Ed., 2010, 49, 6974–6978 CrossRef CAS PubMed
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485–495 CrossRef CAS PubMed
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterization, additional 1H-NMR spectra, absorption and emission data, AFM images and cyclic voltammetry, J/V and PESA plots. See DOI: 10.1039/c3cc43841d |
| This journal is © The Royal Society of Chemistry 2013 |