 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting dense shell/packing principles to invoke stereoselectivity in a reaction accelerated by a chiral dendrimer†
Adam
Ellis
,
Melanie
Wallace
and
Lance J.
Twyman
*
Department of Chemistry, The University of Sheffield, Sheffield, S2 7HF, UK. E-mail: l.j.twyman@sheffield.ac.uk; Fax: +44 (0)114 2229346; Tel: +44 (0)114 2229560
First published on 25th July 2013
Abstract
As dendrimers approach their dense shell or dense packed limit, a certain amount of conformational organization exists. Any substrate binding within the dendrimer's external layer will experience the same organizational effects. This paper describes how these effects can be exploited towards stereocontrol with respect to binding and reactivity.
Our attempts to mimic the reaction rates of enzymes fall into two main classes. The dominant mimetic group consists of small molecules (usually organic soluble), designed to bind substrates and influence reactions through geometric constraints.1 A second approach attempts to mimic the encapsulation behaviour of enzymes using larger polymeric molecules.2,3 This approach enables careful control of internal functionality and more importantly, internal environment. As well as providing specific internal environments, these bulk molecules are often very soluble in water.4 Many types and classes of polymers that have been used to mimic enzyme behaviour. However, the best of these are probably dendrimers.3 These globular polymers possess precisely controlled monodispersed structures. As a result of this control relatively complicated molecules can be created.5
We have previously used the terminal amine groups from a series of dendrimers to accelerate a simple aminolysis reaction in water.6 As part of this study we noticed that the size of the dendrimer had a significant effect on the rate of reaction.7 This was due to the specific encapsulation mechanism of the accelerated reaction,8 which involves hydrophobic binding of the substrate within the outer steric layer of the dendrimer. As such, the substrate is held in close proximity to the terminal (reactive) amine groups. The fastest reaction was observed for the dendrimer with 32 terminal amine groups (3rd generation). Smaller dendrimers with fewer terminal groups possessed structures that were too open, restricting effective binding and geometry. Conversely, the surface amines on larger dendrimers were tightly packed, resulting in congestion and restricted substrate access and binding, leading to much slower reactions. This crowding effect is known as either the dense shell or the dense packed limit.9 Meijer took advantage of this property in an interesting way and designed a dendritic system that could trap molecules internally as its synthesis (and therefore structure) moved above the dense shell limit.10 For the work described in this paper we wish to exploit the situation in a different way and use a 3rd generation dendrimer just below the onset of a dense-shell/packing limit and study any organizational effects on the reactions between the terminal chiral groups of a dendrimer and the stereoisomers of a chiral substrate. Specifically, we describe the aminolysis of hydrophobic Boc-protected L and D phenyl aniline using a 3rd generation water-soluble dendrimer terminated with nucleophilic L-phenylalanine groups. L-phenyl aniline was chosen as the terminal amino acid as it could offer intramolecular π–π and hydrogen bond interactions. In turn, these could help organize any terminal group interactions and therefore stabilize particular terminal conformations.
To test this theory we selected a 2nd generation polypropylenimine, or diaminobutane cored dendrimer (DAB) with 16 terminal groups. Although this is smaller than the optimum dendrimer used in our previous work, the addition of 16 amino acids adds another layer to the periphery of the dendrimer; in effect converting it to the 3rd generation. As such, the surface packing and internal sterics would be similar to the larger/higher generation dendrimer used previously (i.e. with 32 terminal groups).7,8 Peripheral functionalization of the DAB dendrimer with L-phenylalanine was performed in two steps as shown in Scheme 1. The DAB-Am-16 dendrimer 1 was reacted with a 10% excess (per terminal amine) of the Fmoc-L-phenylalanine N-hydroxysuccinimide,11 to produce a protected amino acid terminated DAB dendrimer. The Fmoc protecting groups were removed giving the desired chiral DAB dendrimer 2,12 in a reasonable 27% overall yield. Mass spectrometry, along with 1H and 13C NMR confirmed that reactions had taken place with complete saturation of the terminal groups. Specifically, the MALDI-TOF mass spectrum of the Fmoc protected intermediate generated a molecular ion peak at 7598 (M+), which corresponds to a DAB dendrimer with 16 terminal Fmoc groups. Similarly, the 1H and 13C NMR spectra of the dendrimers confirmed aromatic functionality on the intermediate and final structures. Comparisons of the aromatic and aliphatic integration ratios confirmed the correct number of protons for the 16 terminal Fmoc groups in the intermediate, as well as the 16 terminal benzyl groups on the DAB-AM-16 L-phenylalanine terminated dendrimer 2.
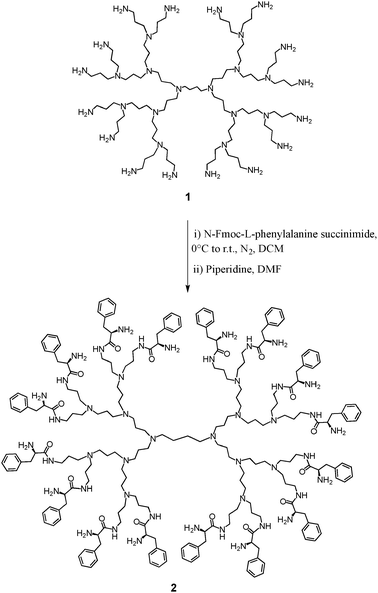 | ||
| Scheme 1 Synthesis of chiral dendrimer 2. | ||
The next step was to select the substrates. In an effort to maximize host-substrate interactions, the p-nitrophenyl active esters of Boc protected L- and D-phenylalanine were chosen (compounds 4 and 5 respectively) (Scheme 2). The functional groups match the terminal amino acid functionalities on the dendrimer. In aqueous solution, any hydrophobically driven interpenetration or surface complex will be accompanied by intermolecular hydrogen bonds and π–π stacking interactions, leading to the formation of a diastereotopic complex, with the polar NO2 groups orientating themselves towards the bulk aqueous solvent. When the L substrate 4 is used, a matched isomeric complex can form between the L terminated dendrimer and the L substrate. Alternatively, a mismatched isomeric complex forms when substrate 5 is used (the L dendrimer and the D substrate). After substrate penetration and complex formation, the dendrimer's terminal amine groups are then free to attack the substrate's active ester and for each reaction two energetically different transition states and products are possible (the L,L or L,D; products 6 and 8 respectively). Due to the chirality on the dendrimers terminal groups, these combined interactions may be optimized for one particular diastereotopic pair, leading to a faster (and selective) aminolysis reaction.
 | ||
| Scheme 2 Aminolysis reaction with dendrimer 2 or the control amine 3. | ||
The reactions were carried out as previously reported;8 with reactions performed at pH 8.5 using the L-phenylalanine terminated DAB-Am-16 2, and either the L or D substrate (compounds 4 and 5 respectively). For all reactions, p-nitrophenolate is liberated during aminolysis,13 which is UV active and enables the concentration of product to be calculated using UV spectrophotometry (by following the increase in the signal intensity at 410 nm).6–8 Our first control experiment simply measured the background reaction of the substrates in buffer (6.0 × 10−5 M solution of substrate 4 or 5 in 0.1 M, pH 8.5 Tris). The initial rate was determined as 3.11 × 10−9 M s−1 and was subsequently subtracted from all future (aminolysis) experiments. A second set of controls designed to measure any difference in rate between the D/L substrates and the chiral amine L-phenylalanine methylester (4, 5 and 3 respectively). The amino acid 3 is an analogue of the dendrimers terminal groups and represents a useful control for the non-encapsulated reactions. Specifically, a 6.4 mM solution of L-phenylalanine methylester 3 was prepared in Tris buffer (0.1 M, pH 8.5). Separate concentrated acetonitrile solutions were made up for the D and the L substrates (compounds 4 and 5 respectively). The D and the L reactions were studied independently by placing a solution of dendrimer 2 into a 1 cm UV cuvette and inserting this into the UV machine. A small aliquot of the concentrated D or the L substrate solution was then added (giving a final substrate concentration of 6.0 × 10−5 M in the cuvette) and the absorption intensity at 410 nm recorded every 10 seconds for a total of 1200 seconds. The experiments were repeated six times. The absorption data was then averaged and plotted against time. The plots were linear over the first 60 seconds and the gradients used to calculate the initial rates for the D and the L substrate (after subtracting the initial rate of the background hydrolysis reaction), Table 1. The rate for the L substrate 4 was slightly faster than the corresponding reaction with the D substrate 5. Although these rates are within the error range of the experiments, all six sets of reactions showed a similar preference for the L substrate. As such we can conclude that the fastest control reaction occurs between the L amino acid 3 and the L substrate 4 (via a lower energy diastereomeric transition state).
| Reaction | Initial rate v0 (M s−1) | Relative ratea | v 0 L relative to v0Db (%) |
|---|---|---|---|
| a v 0/(v0 control 3 + D). b For the control and dendrimer systems respectively = 100 × ((v0L − v0D)/v0D). | |||
| Control 3 + D substrate 5 | 3.71 × 10−9 (±0.45 × 10−9) | 1.00 | 19 |
| Control 3 + L substrate 4 | 4.41 × 10−9 (±0.42 × 10−9) | 1.19 | |
| Dendrimer 2 + D substrate 5 | 3.77 × 10−7 (±0.42 × 10−8) | 101.9 | 61 |
| Dendrimer 2 + L substrate 4 | 6.06 × 10−7 (±0.42 × 10−8) | 163.7 | |
Having carried out the controls and determined the reactive pair preferences, we then proceeded to react the same D and L substrates (4 and 5 respectively) with the L-amino acid dendrimer 2. The reactions were carried out in the same way, except that a 0.4 mM solution of L-phenyl terminated dendrimer 2 was used (giving the same 6.4 mM amine concentration used in the controls). On this occasion the initial rate for the matched system (the L substrate 4 and L terminated dendrimer 2) was much faster than the mismatched pair (the D substrate 5 and L terminated dendrimer 2) – Table 1. If we look at the dendrimer data in more detail and compare the rates to those obtained for the control reactions, we observe relative rate accelerations of 101.9 and 163.7 for the D and the L substrates respectively (when compared to the slowest control reaction), Table 1. These rate accelerations are similar to those previously reported using the optimized 3rd generation non-chiral dendrimers; whose structures were just below the dense packing/dense shell limit.6,7 As with the previous work, the rate accelerations are attributed to the static micellar structure of the dendrimer and the increased solubility of the substrates within the hydrophobic external layer. This places the active ester in close proximity to the outer reactive amines, resulting in an increased local concentration of the reactive partners, leading to a substantial increase in rate. Table 1 also shows the differences in the initial rate for the D and L substrates after reaction with either the single amino acid 3 or the multi amino acid terminated dendrimer 2. It is clear that there is a significant increase in selectivity for the dendrimer-based reaction. Specifically, the initial rate for the aminolysis reaction is 61% faster when the L substrate reacts with the amino acids on the dendrimer's periphery. This compares to a modest 10% increases for the control reaction.
We suggest that the increased selectivity is due to the onset of a dense shell structure. At this point the dendrimer's terminal groups will be able to interact with each other and adopt a minimum energy conformation. Due to the relative proximity and chiral nature of the terminal groups, it is sensible to predict a conformation possessing a certain amount of organizational symmetry.14 Therefore, when the hydrophobic substrates are added to an aqueous solution of the dendrimer, it is equally sensible to suggest that hydrophobic binding will occur in a similarly organized manner (via interpenetration at the surface). As such, it will be energetically more favourable for one enantiomer to bind with respect to the other, leading to a faster reaction. A similar argument can be made for the energy of the diastereomeric transition states.15 In our case, the functionalized dendrimer exhibited a credible degree of diastereoselectivity in favour of the matched system, involving the L-dendrimer 2 and the L-substrate 4.
In conclusion we have presented the synthesis of a chiral dendrimer possessing 16 terminal amines. This dendrimer is capable of accelerating the aminolysis reaction between its terminal amine groups and the active esters of D and L phenyl aniline.13 Furthermore, the dendrimer displayed chiral selectivity with respect to the initial rates of reaction, with the L-substrate reacting 61% faster than the D stereoisomer. The increased rate and chiral selectivity are a result of steric crowding just below the dense shell/packing limit, leading to an organized and low energy diastereomeric conformation.
We would like to thank the E.P.S.R.C. and the University of Sheffield for funding this work.
Notes and references
- (a) G. Parkin, Chem. Rev., 2004, 104(2), 699 CrossRef CAS; (b) S. Julia, J. Masana and J. C. Vega, Angew. Chem., Int. Ed. Engl., 1980, 19(11), 929 CrossRef.
- (a) G. Wulff, T. Gross and R. Schonfeld, Angew. Chem., Int. Ed. Engl., 1997, 36(18), 1962 CrossRef CAS; (b) M. Emgenbroich and G. Wulff, Chem.–Eur. J., 2003, 9, 4108 CrossRef.
- A. Haimov, H. Cohen and R. Neumann, J. Am. Chem. Soc., 2004, 126, 11762 CrossRef CAS; I. M. Klotz, G. P. Roye and I. S. Scarpa, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 263 CrossRef.
- L. J. Twyman, A. S. H. King and I. K. Martin, Chem. Soc. Rev., 2002, 31, 69 RSC.
- (a) G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendritic Molecules, VCH, Weinheim, 1996 Search PubMed; (b) L. Zhiwei and L. Jiangsheng, Curr. Org. Chem., 2013, 17, 1334 CrossRef; (c) J. Hu, T. Xu and Y. Cheng, Chem. Rev., 2012, 112(7), 3856 CrossRef CAS.
- I. K. Martin and L. J. Twyman, Tetrahedron Lett., 2001, 42, 1123 CrossRef CAS.
- J. L. Burnett, A. S. H. King, I. K. Martin and L. J. Twyman, Tetrahedron Lett., 2002, 43, 2431 CrossRef CAS.
- J. L. Burnett, A. S. H. King and L. J. Twyman, React. Funct. Polym., 2006, 66, 187 CrossRef CAS.
- (a) P. G. de Gennes and H. J. Hervet, Phys. Lett., 1983, 44, 351 CrossRef CAS; (b) R. Lescanec and M. Muthukumar, Macromolecules, 1990, 23, 2280 CrossRef CAS; (c) D. Boris and M. Rubinstein, Macromolecules, 1996, 29(22), 7251 CrossRef CAS.
- J. F. G. A. Jansen, E. W. Meijer and E. M. M. de Brabander-van den Berg, J. Am. Chem. Soc., 1995, 117, 4417 CrossRef CAS.
- Functionalisation of all terminal groups requires the use of excess reagents and longer reaction times. For example see: P. Ballester, R. M. Gomila, C. A. Hunter, A. S. H. King and L. J. Twyman, Chem. Commun., 2003, 38 RSC.
- B. Romagnoli and W. Hayes, J. Mater. Chem., 2002, 12, 767–799 RSC.
- Due to the small amounts of starting material we were not able to synthesise the dipeptide products 6 and 7, or to specifically identify them during the aminolysis reaction. However, based on our recent studies using dendrimers (ref. 6 and 7 above) and other work on polymer based aminolysis (see for example: D. J. Evans, A. Kanagasooriam, A. Williams and R. J. Pryce, J. Mol. Catal., 1993, 85(1), 21 CrossRef CAS ), we are confident that aminolysis and not hydrolysis is occurring. More evidence comes from LC-MS, which could not detect any of the N-Boc-phenylaniline during the dendrimer catalysed reactions (i.e. the hydrolysis by-product). Nevertheless, we do acknowledge that the dendrimer reactions maybe more complex and a number of cleavage processes may be occurring/competing; including aminolysis and possibly hydrolysis. Nevertheless, the overall outcome and conclusions remain the same. that is, the dendrimer reactions are rate enhanced and demonstrate chiral selectivity based on a dense shell structure.
- A similar effect has been observed for porphyrin terminated dendrimers interacting with bidentate ligands; J. Yang, S. Cho, H. Yoo, J. Park, W. S. Li, T. Aida and D. J. Kim, Phys. Chem., 2008, 112(30), 6869 CrossRef CAS , and the ordered structure of liquid crystal dendrimers; V. Percec and M. Kawasumi, Macromolecules, 1992, 25(15), 3843 CrossRef.
- (a) C. E. Masse and J. S. Panek, Chem. Rev., 1995, 95(5), 1293 CrossRef CAS; (b) A. Bernardi, A. M. Capelli, A. Comotti, C. Gennari, M. Gardner, J. M. Goodman and I. Paterson, Tetrahedron, 1991, 20, 3471 CrossRef; (c) K. Gothelf, I. Thomsen and K. A. Jørgensen, J. Am. Chem. Soc., 1996, 118(1), 59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, catalysis and control experiments. See DOI: 10.1039/c3cc43830a |
| This journal is © The Royal Society of Chemistry 2013 |