 Open Access Article
Open Access ArticleIsostructural salts of the same complex showing contrasting thermal spin-crossover mediated by multiple phase changes†‡
Thomas D.
Roberts
a,
Marc A.
Little§
a,
Floriana
Tuna
b,
Colin A.
Kilner
a and
Malcolm A.
Halcrow
*a
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, UK LS2 9JT. E-mail: m.a.halcrow@leeds.ac.uk; Fax: +44 (0)113 343 6565; Tel: +44 (0)113 343 6506
bSchool of Chemistry and Photon Science Institute, University of Manchester, Oxford Road, Manchester, UK M13 9PL
First published on 30th May 2013
Abstract
Two salts of [FeL2]2+ (L = 2,6-bis[5-methyl-1H-pyrazol-3-yl]pyridine) are isostructural under ambient conditions but show different thermal spin-crossover behaviour, involving a variety of crystallographic phase changes.
While an increasing number of applications for spin-crossover complexes1–6 in devices and nanoscience has been demonstrated,3 the number of materials showing technologically favourable spin-state switching is still very small.5 There is therefore continued interest in the structural chemistry of spin-transition materials, which ultimately aims to crystal engineer new spin-crossover compounds with pre-defined functionality.6
We recently reported that [FeL2][BF4]2 (1; L = 2,6-bis(5-methyl-1H-pyrazol-3-yl)pyridine) adopts different anhydrous forms under slow crystallisation, and upon thermal dehydration of hydrated crystals.7 The latter material undergoes an abrupt spin-transition around 205 K with wide thermal hysteresis, which is coupled to a sequence of three crystallographic phase changes (see below).7 This behaviour, which was mostly elucidated by X-ray powder diffraction, makes 1 one of the most structurally complex spin-transition compounds known.6 We now describe the corresponding perchlorate salt [FeL2][ClO4]2 (2), which is isostructural with 1 under ambient conditions, but exhibits very different spin-state properties.
As we have previously described, 1 usually crystallises from organic solvents as the brown hydrate 1·2H2O, which contains two unique iron centres.7 One of these is high-spin, and donates N–H⋯F hydrogen bonds to four BF4− anions. The other is low-spin, and forms N–H⋯O interactions to four water molecules. An anhydrous phase of the same compound (phase 1A) can also be obtained, which is structurally similar to 1·2H2O but with the BF4− ions disordered onto the vacant water sites in the lattice. Exposure of 1A to air leads to its rapid hydration to 1·2H2O.
Heating 1·2H2O to 400 K converts it to a different high-spin anhydrous material, 1B, which is distinct from 1A by powder diffraction. Cooling 1Bin vacuo leads to two consecutive phase changes 1B → 1C → 1D near 303 and 270 K respectively, without changing its high-spin state. Phase 1D then undergoes a cooperative transition to a fifth, low-spin phase 1E. This spin-transition is centred near 205 K with a hysteresis loop of 37–65 K, depending on the history of the sample.
Unlike 1, the structural chemistry of 2 is complicated by pseudopolymorphism. Recrystallisation of 2 from organic solvents often leads to mixtures of unsolvated and solvate phases. However, orange-yellow crystals of unsolvated 2 (phase 2A) can be obtained in pure form by diffusion of diethyl ether into dilute methanol solutions of the complex at room temperature (more concentrated solutions instead afford the methanol solvate described below, which can be distinguished from 2A by its brown colouration). Crystalline 2A is isostructural with anhydrous phase 1A although in contrast to 1A, which is low-spin at 150 K, 2A contains a residual high-spin fraction at that temperature by X-ray diffraction (ESI‡). As for 1A, crystals of 2A are hygroscopic and form brown 2·2H2O over a period of days on exposure to air at room temperature. The resultant crystals afford a similar unit cell to 1·2H2O at room temperature.¶![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 Moreover, bulk samples of 1·2H2O and 2·2H2O are isostructural at room temperature by X-ray powder diffraction (ESI‡).
7 Moreover, bulk samples of 1·2H2O and 2·2H2O are isostructural at room temperature by X-ray powder diffraction (ESI‡).
Despite their isostructural nature, the spin-state behaviour of 1·2H2O and 2·2H2O is very different (Fig. 1). Solid 1·2H2O exhibits an almost invariant 1:1 high:low-spin state population between 5–300 K.7 In contrast, freshly prepared 2·2H2O is ca. 85% high-spin at room temperature, according to its χMT value of 3.1 cm3 mol−1 K. This value decreases on cooling, with a small discontinuity around 265 K (Fig. 1). More cooling leads χMT to decrease further in a gradual but irregular manner until 120 K, when the remaining 25% of the sample undergoes a more abrupt spin-conversion, reaching its fully low-spin state at 110 K. Rewarming the sample shows the transition to exhibit a structured hysteresis below 200 K. The hysteresis loop spans 65 K at its widest point, and involves ca. 40% of the iron centres in the material. Repeated thermal cycling between 300–5–300 K caused some changes to the shape of the χMT vs. T curve, although the discontinuity at 265 K and the low-temperature hysteresis were retained.
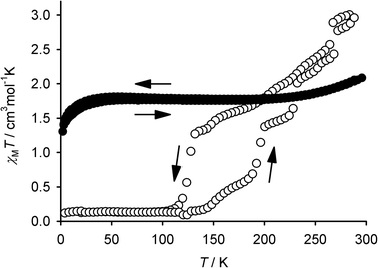 | ||
| Fig. 1 Comparison of the magnetic susceptibility behaviour of 1·2H2O (●)7 and 2·2H2O (○) below room temperature. Both samples were scanned in both cooling and warming mode. | ||
Variable temperature X-ray powder diffraction on 2·2H2O shed some light on these observations (Fig. 2). The discontinuity in χMT at 265 K (Fig. 1) is associated with a crystallographic transformation to a new phase, labelled 2·2H2O* in Fig. 2. This phase change is not exhibited by 1·2H2O.7 The sample retains the 2·2H2O* structure on cooling between 250–130 K, with only minor changes to the powder pattern being observed (Fig. 2). It is unclear whether the more abrupt part of the transition, that gives rise to the bulk of the hysteresis, involves a further crystallographic phase change at around 110 K.¶6–8
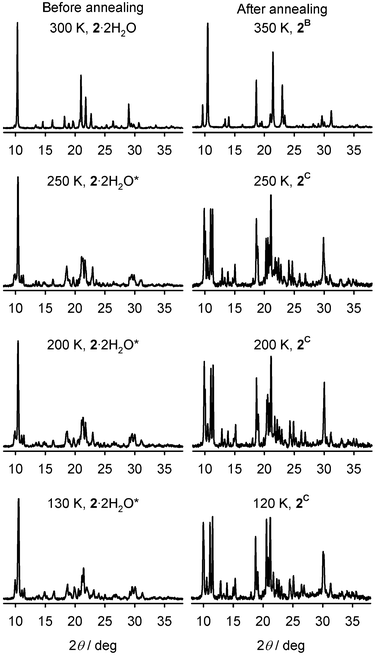 | ||
| Fig. 2 Variable temperature X-ray powder patterns of 2·2H2O before and after annealing at 370 K. The plots are labelled with the phases adopted by the material at each temperature. The powder patterns for 2C are noisier because they were obtained from nujol mull samples in vacuo. | ||
Heating 2·2H2O resulted in the loss of 1.5 equiv. of water at 368 K by thermogravimetric analysis (the highest temperature measured on safety grounds). Annealing 2·2H2O at 370 K leads to its conversion to a new yellow anhydrous phase 2B, which is isostructural with 1B and reverts to brown 2·2H2O on recooling in air. This transformation occurred rapidly in the powder diffractometer, where the sample was directly open to the vacuum (Fig. 2), but was more gradual in the magnetometer (ESI‡). As for 1, the anhydrous phases of 2 produced by slow crystallisation (2A) and thermal dehydration of the dihydrate (2B) are distinct from each other by X-ray powder diffraction.
The variable temperature spin-state properties of phases 2B and 1B are again different. While 1B exhibits a highly cooperative spin transition centred near 205 K,72B remains high-spin upon cooling to 2 K (Fig. 3). In the powder diffractometer, cooling 2B below 295 K caused a transformation to a new phase 2C, which has a very different powder diffraction pattern (Fig. 2). The corresponding material 1B also converts to 1C under these conditions, at the slightly higher temperature of 303 K.7 Phases 2B and 2C are isostructural with 1B and 1C (ESI‡). However, whereas 1C transforms further to 1D on additional cooling, the phase 2C is retained between 120–250 K (Fig. 2 and 3). Hence, 2C (and presumably 1C) are high-spin structures, and spin-crossover in 1 can only occur owing to the extra 1C → 1D phase change.
 | ||
| Fig. 3 Comparison of the magnetic susceptibility behaviour of 1B (◆, top)7 and 2B (⋄, bottom), and their phase changes between 120 and 350 K. The red lines show the temperatures of each phase transition. | ||
In addition to the above, single crystal X-ray analyses were obtained from two solvates of 2. The methanol solvate 2·2CH3OH contains one formula unit per asymmetric unit, whose complex cation is low-spin at 150 K. The other solvate 2·xCH3NO2·⅓(C2H5)2O (x ≈ 0.83) contains three unique molecules in its asymmetric unit, one of which is low-spin at 150 K while the other two have a mixed high/low-spin population. Both structures contain extensive hydrogen bonding between the complex, anions and solvent (ESI‡).
The complex cations in 2·2H2O,¶2A, the corresponding phases of 1 and the solvate structures of 2 all associate into four-fold “terpyridine embrace” layers, through interdigitation of the pyrazolyl arms of neighbouring molecules.9,10 These cation layers are separated in the lattice by sheets of anions and, where present, solvent. The cation layers in 2A and 2·2H2O are homochiral and contain strictly or approximately co-aligned complex molecules, with adjacent layers being related by a crystallographic inversion centre. In contrast, the layers in the solvate crystals contain molecules of both handedness, which are canted with respect to each other. The interplanar distance between adjacent, overlapping pyrazole rings in the layers varies from 3.3–3.9 Å in the four materials. Despite their individual differences, 1 and 2 clearly show a strong preference for this layered structure type, which allows some flexibility in the orientation of the molecules. Hence, we suggest that the multiple phase changes in 1, 2 and their hydrate materials might reflect facile, reversible canting of the cations in the terpyridine embrace layers, coupled to movement or (dis)ordering of the anions in the space between those layers. Such rearrangements could also lead to changes to the hydrogen bonding at the [FeL2]2+ cations, which would further influence their spin states in the different phases.11
In conclusion, the complicated structural and spin-state chemistry of the [FeL2]2+ system is even more complex than first reported.7 The BF4− and ClO4− salts form dihydrate and anhydrous phases which are isostructural, but show contrasting spin-state behaviour. Thus, 2·2H2O exhibits thermal spin-crossover but 1·2H2O does not. Conversely, anhydrous 2B is not spin-crossover active, while 1B exhibits a highly cooperative spin-transition below room temperature. X-ray powder diffraction has demonstrated that these results reflect different sequences of phase transitions in the two sets of materials. Calorimetry and Mössbauer spectroscopic studies are in progress to shed more light on this unprecedented structural chemistry.
The authors thank Dr Tamsin Malkin (University of Leeds) for help with the powder diffraction data. This work was funded by the EPSRC, the University of Manchester and the University of Leeds.
Notes and references
- Spin Crossover in Transition Metal Compounds I–III, Top. Curr. Chem., ed. P. Gütlich and H. A. Goodwin, 2004, vol. 233–235 Search PubMed.
- Spin-crossover materials – properties and applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013, p. 568 Search PubMed.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313 RSC.
- For recent reviews see: M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS; P. Gamez, J. S. Costa, M. Quesada and G. Aromí, Dalton Trans., 2009, 7845 RSC; J. Tao, R.-J. Wei, R.-B. Huang and L.-S. Zheng, Chem. Soc. Rev., 2012, 41, 703 RSC; P. Gütlich, Eur. J. Inorg. Chem., 2013, 581 CrossRef.
- I. Šalitroš, N. T. Madhu, R. Boča, J. Pavlik and M. Ruben, Monatsh. Chem., 2009, 140, 695 CrossRef.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119 RSC.
- T. D. Roberts, F. Tuna, T. L. Malkin, C. A. Kilner and M. A. Halcrow, Chem. Sci., 2012, 3, 349 RSC.
- Other examples of strongly hysteretic spin-crossover, whose structural chemistry is well understood, include: J.-F. Létard, P. Guionneau, E. Codjovi, O. Lavastre, G. Bravic, D. Chasseau and O. Kahn, J. Am. Chem. Soc., 1997, 119, 10861 CrossRef CAS; M. Nihei, H. Tahira, N. Takahashi, Y. Otake, Y. Yamamura, K. Saito and H. Oshio, J. Am. Chem. Soc., 2010, 132, 3553 CrossRef; G. A. Craig, J. S. Costa, O. Roubeau, S. J. Teat and G. Aromí, Chem.–Eur. J., 2011, 17, 3120 CrossRef; H. J. Shepherd, T. Palamarciuc, P. Rosa, P. Guionneau, G. Molnár, J.-F. Létard and A. Bousseksou, Angew. Chem., Int. Ed., 2012, 51, 3910 CrossRef.
- I. Dance and M. Scudder, CrystEngComm, 2009, 11, 2233 RSC.
- R. Pritchard, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2007, 577 RSC.
- S. A. Barrett, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2011, 40, 12021 RSC and refs. therein.
Footnotes |
| † Celebrating 300 years of Chemistry at Edinburgh. |
| ‡ Electronic Supplementary Information (ESI) available: Experimental descriptions, crystallographic figures and tables, and additional powder diffraction data. CCDC 936803–936805. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc43613f |
| § Current address: Department of Chemistry, University of Liverpool, Crown Street, Liverpool, UK L69 7ZD. |
| ¶ X-ray diffraction data were collected from the same single crystal of 2·2H2O at 296, 200 and 105 K. At 296 K the data were solved in I41/a, the space group adopted by 1·2H2O, but refined poorly (crystals of 1·2H2O produced by hydration of preformed 1A show similar behaviour7). At the lower temperatures good quality refinements were achieved, but it was not possible to unambiguously assign a space group to the material. Despite these ambiguities, these crystallographic refinements confirm that the disposition of the molecules in 2·2H2O is identical to that in 1·2H2O.7 They are also consistent with the magnetic susceptibility data in showing that 2·2H2O has an approximate 1:1 high:low-spin population at 200 K, and is fully low-spin at 105 K. More details are given in the ESI.‡ |
| This journal is © The Royal Society of Chemistry 2013 |