 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly selective route to linear alpha olefins from biomass-derived lactones and unsaturated acids†
Dong
Wang
,
Sikander H.
Hakim
,
David
Martin Alonso
and
James A.
Dumesic
*
Department of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA. E-mail: dumesic@engr.wisc.edu
First published on 27th June 2013
Abstract
This work demonstrates the use of Lewis-acid catalysts, such as gamma-alumina and tungstated alumina, for selective production of linear alpha olefins by decarboxylation of lactones and unsaturated carboxylic acids.
The use of biomass feedstocks is central to satisfy the long-term need for carbon-based commodity chemicals in a sustainable manner. A promising strategy for the conversion of biomass-derived carbohydrates to value-added chemicals is to take advantage of synergies between the high chemo- and stereo-selectivity of biocatalysis and the high efficiency of chemical catalysis.1 An example is polyketide/fatty acid biosynthesis, which offers flexibility to produce targeted molecules with specified carbon chain lengths and functionalities within a homologous series based on a single metabolic pathway.2 The products assembled through this route create a diverse platform, which can be subsequently upgraded by chemical catalysis to access families of commodity chemicals. Herein, we present a process that can decarboxylate unsaturated carboxylic acids and lactones (Cn) obtained by the above biological routes to selectively produce linear alpha olefins (LAOs) (Cn−1), which comprise a family of valuable commodity chemicals with versatile applications as polymers, detergents and other specialty chemicals.3 This strategy uses inexpensive, robust heterogeneous solid acid catalysts, and it is suitable for a range of unsaturated carboxylic acids and their lactones.
The production of LAOs is inherently challenging due to the ease of migration of the terminal double bond to internal positions. LAOs are manufactured commercially from fossil fuel feedstocks by two main routes, oligomerization of ethylene and cracking of Fischer–Tropsch products.4,5 These processes necessarily lead to a distribution of LAOs with different chain lengths. Moreover, LAOs with odd carbon numbers cannot be produced by direct ethylene oligomerization. In addition, current processes overwhelmingly rely on fossil carbon and thus are not sustainable. In this regard, the development of processes for production of LAOs from renewable feedstocks has become of wide interest. For instance, a process has been demonstrated for LAO production by decarbonylation–dehydration of fatty acids over homogenous catalysts based on Pd.6 Another process involves silver(II) mediated decarboxylation of fatty acids followed by ethenolysis over a commercial metathesis catalyst to produce renewable LAOs.7
We have recently reported a novel route to produce olefins from renewable resources by solid-acid catalyzed decarboxylation reaction of biomass-derived lactones and unsaturated carboxylic acids.8–10 When an aqueous solution of γ-valerolactone (GVL) is reacted over a solid acid catalyst such as silica–alumina (SiO2–Al2O3) at temperatures near 648 K, the GVL reactant undergoes ring-opening to produce 3- and 4-pentenoic acids, which then isomerize to 2-pentenoic acid (Scheme 1). 2-Pentenoic acid then irreversibly decarboxylates to form 1-butene and an equimolar amount of carbon dioxide, likely via a β-carbenium ion intermediate.9 The 1-butene product then undergoes rapid isomerization over the SiO2–Al2O3 catalyst to form trans- and cis-2-butene. Under typical operating conditions used to maximize the total butene yield from GVL (e.g., 30–60 wt% GVL in water, 648 K, 1 bar, WHSV = 0.18–0.90 h−1),8 the isomerization of 1-butene essentially proceeds to equilibrium, and only 23% of the C4 stream is retained as 1-butene.11 Therefore, it is challenging to selectively produce 1-butene or other α-olefins using our previously-studied solid Brønsted acid catalysts such as SiO2–Al2O3, because these catalysts exhibit high activity for both formation of 1-butene and its isomerization. Nevertheless, the decarboxylation of γ-lactones and the corresponding olefinic acids could provide a new route to terminal olefins, provided that the olefin isomerization reaction can be effectively suppressed.
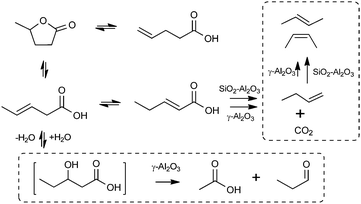 | ||
| Scheme 1 Decarboxylation of γ-valerolactone. | ||
In this work, we show that a stream of C4 olefin containing unprecedentedly high levels of 1-butene can be produced in a single flow reactor when a Lewis acid catalyst, such as γ-Al2O3, is used to catalyze the decarboxylation of GVL (Fig. 1, right). Using this catalyst, nearly pure 1-butene (>99% of C4 stream) can be obtained (10% total butene yield). Increasing the space time increases the yield to total butenes, which also leads to a slight decrease in percentage of 1-butene in the butene product stream. However, over 92% selectivity to 1-butene can be achieved at 43% total butene yield. In contrast to the desirable performance of the γ-Al2O3 catalyst, the Brønsted acid SiO2–Al2O3 catalyst that we had previously found to afford the highest yield to total butene from GVL, showed only 24% selectivity to 1-butene at 90% total butene yield. After optimizing the reaction conditions (Fig. 1, left), 92% of the product olefin stream can be retained as 1-butene using SiO2–Al2O3 as catalyst, but only at 2% total butene yield. Increasing the total olefin yield to 30% decreased the 1-butene percentage to 72%. Given the trade-off between 1-butene selectivity and total butene yield over SiO2–Al2O3, the maximum yield to 1-butene is limited to approximately 22%. If a stream of highly pure 1-butene (e.g. >90%) is desired, the reactor needs to be operated at very low conversion levels, requiring separation of the butene product from GVL, and recycle of unreacted GVL to the reactor inlet.
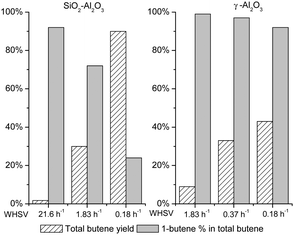 | ||
| Fig. 1 Butene production by decarboxylation of GVL over SiO2–Al2O3 and γ-Al2O3 catalysts. 30 wt% GVL in water, 648 K, 1 bar. | ||
It is well-established that γ-Al2O3 possesses predominantly Lewis acid sites, while mixed oxides such as SiO2–Al2O3, possesses both Brønsted and Lewis acid sites.12 The higher selectivity for production of 1-butene over γ-Al2O3 thus indicates that Lewis acid sites effectively catalyze GVL decarboxylation to produce 1-butene, but they exhibit significantly lower rates for isomerization of 1-butene. To explore further the active sites on this catalyst, γ-Al2O3 was modified with varying loadings of tungsten oxide (WOx). Table 1 compiles the physiochemical properties and catalytic activities for butene production over selected solid catalysts at high conversions of GVL. The densities of Brønsted and Lewis sites were measured by a combination of NH3 temperature programmed desorption (TPD) (total number of acid sites) and infrared spectroscopy of adsorbed pyridine (ratio of Brønsted and Lewis sites). It can be seen (entries 4–6) that increasing the loading of WOx from 0 to 20 wt% progressively creates Brønsted acidity at the expense of Lewis acidity, consistent with a previous report by Stoled et al.13 Consequently, the catalytic behaviour of WOx–Al2O3 with increasing WOx loading should approach that of SiO2–Al2O3 (entry 1), whose activity is dominated by Brønsted acidity. This expected trend is clearly evidenced by the low 1-butene selectivity observed at high WOx loading (entry 6). Remarkably, 4 wt% WOx–Al2O3 catalyst is able to retain 92% of total C4 olefin as 1-butene at total butene yield of 70%, leading to 64% yield to 1-butene (entry 4).
| Entry | Catalyst | CO2 uptake (μmol g−1) | BASc/LAS (μmol g−1) | GVL conv. (%) | C=4 yieldd (%) | AAe yield (%) | PEAf yield (%) | 1-C=4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g (%) g (%) |
|---|---|---|---|---|---|---|---|---|
| a Reaction T = 723 K. b Denotes 4 wt% WOx–Al2O3. c Brønsted acid sites/Lewis acid sites. d Denotes butene. e Denotes acetic acid. f Denotes pentenoic acid. g 1-Butene percentage in total butenes. | ||||||||
| 1 | SiO2–Al2O3 | <1 | 546/136 | 98 | 92 | <1 | 3 | 24.2 |
| 2 | γ-Al2O3 | 55 | 0/219 | 97 | 43 | 16 | 1 | 92.2 |
| 3 | γ-Al2O3a | 55 | 0/219 | 99 | 59 | 8 | 2 | 82.4 |
| 4 | 4W-Alb | 27 | 5/230 | 98 | 70 | 10 | 1 | 91.8 |
| 5 | 8W-Al | 10 | 32/191 | 96 | 73 | 8 | 3 | 71.0 |
| 6 | 20W-Al | 7 | 141/107 | 95 | 80 | 3 | 5 | 27.9 |
| 7 | MgO | 220 | — | 83 | 2 | 15 | 22 | 88.9 |
We next demonstrate that higher LAOs can also be accessed by decarboxylation of lactones with longer carbon chains, which can be derived from the polyketide/fatty acid synthesis. It is shown in Table 2 that γ-hexalactone (GHL), γ-octalactone (GOL) and γ-undecalactone (GUL) can be converted over 4 wt% WOx–Al2O3 to form 1-pentene, 1-heptene and 1-decene, respectively. Remarkably high LAO percentages (>95%) can be obtained at moderate total olefin yields for the C6 and C8 lactones (entries 1 and 3), although at higher total olefin yields (entries 2 and 4), the LAO percentages decrease below 90%. We observe that maintaining a high LAO percentage at a high total olefin yield becomes challenging with GUL (entries 5 and 6), whereas a high 1-decene percentage can be obtained at lower total decene yield.
| Entry | Feeda | Lactone conv. (%) | Olefin yield (%) | AAd yield (%) | Unsaturated acids yield (%) | LAOe (%) |
|---|---|---|---|---|---|---|
| a Co-fed with water using two syringe pumps. b Reaction T = 648 K. c Reaction T = 673 K. d Denotes acetic acid. e LAO percentage in total olefins. | ||||||
| 1 | 5 wt% GHLb | 82 | 54 | 8 | 12 | 97.1 |
| 2 | 5 wt% GHLb | 98 | 61 | 13 | 2 | 88.3 |
| 3 | 30 wt% GOLc | 91 | 16 | <1 | 58 | 96.5 |
| 4 | 10 wt% GOLc | 97 | 58 | 2 | 16 | 80.9 |
| 5 | 30 wt% GULc | 88 | 7 | 4 | 56 | 96.8 |
| 6 | 10 wt% GULc | 97 | 17 | 5 | 40 | 85.7 |
In addition to the desirable decarboxylation reaction, we have also observed side reactions during conversion of GVL over γ-Al2O3. In particular, equimolar amounts of acetic acid and propionaldehyde are formed as major by-products, with yields up to 16% of total GVL converted. Notably, acetic acid and aldehydes with two-fewer carbon atoms compared to the lactone reactant (e.g., hexanaldehyde for GOL) are produced as major by-products, regardless of the nature of the starting lactones. This behaviour strongly suggests that acetic acid is likely formed via a retro-aldol type mechanism through a common 3-hydroxy acid intermediate for different lactones, such as the 3-hydroxypentanoic acid in the case of GVL (Scheme 1). Also, a portion of carbon goes to unidentified condensation products, likely formed between butene and GVL or pentenoic acids over γ-Al2O3. It appears that higher lactones and acids have lower reactivity towards decarboxylation and are more prone to side reactions. Indeed, thermogravimetric analysis reveals that for the same mass of lactones processed, 50% more carbon is deposited on catalysts used to process GUL than used for GHL. Thus, further optimization is needed to improve olefin yield from these feedstocks.
Table 1 summarizes basic site densities as titrated by CO2-TPD. The extent of CO2 uptake is negligible for SiO2–Al2O3, whereas the CO2 uptake for γ-Al2O3 is 55 μmol CO2 g−1. Accordingly, we suggest that the formation of acetic acid and propionaldehyde on the γ-Al2O3 catalyst takes place by a retro-aldol type mechanism, which is known to be catalyzed by basic sites.14 To test this hypothesis, we studied the decarboxylation of GVL over magnesium oxide (MgO), a basic oxide. The MgO catalyst produced over seven times more acetic acid than butene from GVL conversion, implicating the importance of basic sites in this pathway. Importantly, the basic site density of γ-Al2O3 can be decreased by the progressive addition of WOx (0 to 20 wt%), leading to a corresponding decrease in the yields to acetic acid and condensation products (Table 1). In this respect, we suggest that future research should explore the synthesis of water-tolerant solid acid catalysts bearing predominantly Lewis acidity with minimal basicity, which would be ideal for the selective production of LAOs via decarboxylation.
It is interesting (and unexpected) that γ-Al2O3 and WOx–Al2O3 can be used to selectively produce LAOs since both catalysts are known to have good activity for olefin double bond migration and skeletal isomerization at temperatures similar to those used in the present study (673–800 K).15 The key discovery of the present work, however, is that the presence of water significantly inhibits the undesirable isomerization of the alpha-olefins over Lewis acid sites, whereas this isomerization is rapid over Brønsted acid sites, even in the presence of water. For example, the steady-state rates of 2-pentenoic acid decarboxylation and 1-butene isomerization over γ-Al2O3 are 0.012 (feed: Ptotal = 1 bar, PPEA = 0.07 bar, helium balance) and 0.15 mmol min−1 gcat−1 (Ptotal = 1 bar, P1-butene = 0.005 bar, helium balance), respectively. However, when water was co-fed, the decarboxylation rate increased to 0.051 (Ptotal = 1 bar, PPEA = 0.07 bar, Pwater = 0.93 bar) while the 1-butene isomerization rate decreased to 0.020 mmol min−1 gcat−1 (Ptotal = 1 bar, P1-butene = 0.005 bar, Pwater = 0.90 bar, helium balance). Under the same conditions, the rates of decarboxylation and 1-butene isomerization over SiO2–Al2O3 without water are 0.48 and 8.78 mmol min−1 gcat−1, respectively. When water was admitted into the system, the decarboxylation rate increased to 0.52 while the isomerization rate decreased only moderately to 4.50 mmol min−1 gcat−1. Hence, adding water decreases the 1-butene isomerization rate by a factor of 7.5 over γ-Al2O3 and 1.9 over SiO2–Al2O3. Our finding thus represents an interesting case where the high water content commonly encountered in biomass processing can be utilized to positively influence product selectivity. The origin of high 1-butene selectivity over these catalysts, as well as the detailed effect of water on the product distribution, are currently under investigation and will be reported in a following paper.
In summary, we demonstrated that it is possible to produce a stream of highly pure LAOs from lactones or unsaturated carboxylic acids in a single step using inexpensive, robust heterogeneous acid catalysts with no precious metal components. We show that analogous decarboxylation chemistry exists for C5 C6, C8 and C11 γ-lactones, which indicates the potential of this methodology as a general approach for the production of LAOs with varying chain lengths. The feedstock to this process can be derived from biological routes (e.g., polyketide/fatty acid biosynthesis), which thereby generates a series of homologous molecules for conversion to LAOs. Moreover, unlike the present ethylene-based route for production of LAOs, this new biological route is capable of targeting molecules with specified carbon chain length which after subsequent decarboxylation with the current technology, leads to a single LAO cut, thus offering additional manufacturing flexibility to meet market demand. This approach also allows for the production of both even and odd carbon number LAOs, depending on the number of carbons in the starting molecules. Such an integrated approach demonstrates the promise of a synergy between biocatalysis and chemical catalysis for efficiently converting biomass into value-added chemicals.
This work was supported in part by the U.S. Department of Energy Office of Basic Energy Sciences and by the DOE Great Lakes Bioenergy Research Center, which is supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through Cooperative Agreement between the Board of Regents of the University of Wisconsin System and the U.S. Department of Energy. In addition, this material is based upon work supported by the National Science Foundation under Award No. EEC-0813570.
Notes and references
- B. H. Shanks, ACS Chem. Biol., 2007, 2, 533–535 CrossRef CAS.
- B. J. Nikolau, M. A. D. N. Perera, L. Brachova and B. Shanks, Plant J., 2008, 54, 536–545 CrossRef CAS.
- D. Vogt, in Applied Homogeneous Catalysis With Organometallic Compounds, 1996, vol. 1, pp. 245–258 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, 3rd edn, 1997, ch. 3, pp. 75–83 Search PubMed.
- Alpha Olefins(02/03-4) PERP Report, Nexant Chemical Systems, 2004.
- G. A. Kraus and S. Riley, Synthesis, 2012, 3003–3005 CrossRef CAS.
- F. van der Klis, J. Le Nôtre, R. Blaauw, J. van Haveren and D. S. van-Es, Eur. J. Lipid Sci. Technol., 2012, 114, 911–918 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- J. Q. Bond, D. Wang, D. M. Alonso and J. A. Dumesic, J. Catal., 2011, 281, 290–299 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, R. M. West and J. A. Dumesic, Langmuir, 2010, 26, 16291–16298 CrossRef CAS.
- H. H. Voge and N. C. May, J. Am. Chem. Soc., 1946, 68, 550–553 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- S. L. Soled, G. B. McVicker, L. L. Murrell, L. G. Sherman, N. C. Dispenziere Jr, S. L. Hsu and D. Waldman, J. Catal., 1988, 111, 286–295 CrossRef CAS.
- R. Mahrwald, Modern Aldol Reactions, Wiley-VCH, 2004 Search PubMed.
- L. H. Gielgens, M. G. H. Vankampen, M. M. Broek, R. Vanhardeveld and V. Ponec, J. Catal., 1995, 154, 201–207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc43587c |
| This journal is © The Royal Society of Chemistry 2013 |