 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical and magnetic properties of a surface-grafted novel endohedral metallofullerene derivative†‡
Núria
Crivillers
a,
Yuta
Takano
bc,
Yuya
Matsumoto
b,
Javier
Casado-Montenegro
a,
Marta
Mas-Torrent
a,
Concepció
Rovira
a,
Takeshi
Akasaka
*bd and
Jaume
Veciana
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC) and CIBER-BBN, Campus de la UAB, 08193, Bellaterra, Spain. E-mail: vecianaj@icmab.es
bLife Science Center of Tsukuba Advanced Research Alliance, University of Tsukuba, Tsukuba, Ibaraki 305-8577, Japan. E-mail: akasaka@tara.tsukuba.ac.jp
cInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
dFoundation for Advancement of International Science (FAIS), Tsukuba, Ibaraki 305-0821, Japan
First published on 28th June 2013
Abstract
A novel endohedral metallofullerene (EMF) has been designed and synthesised for its grafting on gold. The functionalization of the substrate resulted in a multifunctional surface displaying the properties of the EMF in solution.
Endohedral metallofullerenes (EMFs)1 are very attractive systems because of their unique optical, magnetic and electronic properties differing completely from those of the empty fullerenes.2 Indeed, these derivatives exhibit the combined properties of the individual components along with additional properties appearing from the metal–cage interaction. These characteristics make them very appealing for a variety of applications in fields ranging from molecular electronics to nanomedicine.2a However, for their future implementation in devices, the controlled deposition and immobilization of these functional molecules on certain substrates is demanded. The deposition by physisorption of M@C82 (e.g. M = La, Ce, Tb) on different surfaces has been investigated, commonly by means of Scanning Tunneling Microscopy (STM).2c,3 Nonetheless, the fabrication of more robust hybrid surfaces requires to chemically bond the molecules on the substrates and, thus, derivatives with a suitable grafting group might be prepared. Due to the difficulty in isolating the pure isomers in reasonable amounts and their synthetic complexity, to our knowledge, there is only one reported example regarding the chemisorption of EMFs on surfaces. This was carried out with a trimetallic nitride templated EMF (TNT-EMF), Er3N@C80, functionalized with a dithiolane group, and it was demonstrated that once deposited on gold its luminescence was not quenched.4 Striking differences in the electronic properties exist between TNT-EMFs and La@C82 led by an electron transfer from the inside metal to the fullerene cage in the latter. As a result, La@C82 has remarkably low oxidation and high reduction potentials (Ered1: −0.42 V, Eox1: 0.07 V. cf. Er3N@C80; Ered1: −1.42 V, Eox1: 0.63 V, vs. ferrocene0/+),5 and a unique open shell structure of the fullerene cage.5,6 Such electronic nature is much more attractive for the development of novel molecular materials than that of other EMFs.7
Here, for the first time, a novel La@C82 derivative, functionalized with a thioacetyl protecting anchoring group to be chemisorbed on gold has been successfully synthesized and employed for preparing self-assembled monolayers (SAMs). We demonstrate that the electrochemical and magnetic properties of the novel EMF can be transferred and preserved upon its chemisorption on gold. In addition, the outstanding redox response of the SAM has permitted to electrically trigger its redox state and consequently its magnetic behavior. These results suggest that these SAMs are promising electrochemical molecular switches in which their magnetic properties can be exploited as the output signal.
The target La@C82 derivative (2) was synthesized by 1,3-dipolar cycloaddition reaction using a thioacetate-terminated aldehyde (1) (Scheme 1). Utilizing 2-methylaminoisobutyric acid in the present reaction afforded one major regioisomer (2b) selectively, in contrast with the reactions using N-methylglycine or N-n-octylglycine instead of aminoisobutyric acid.8 In the HPLC profile of the reaction mixture, sharp peaks corresponding to two regioisomers (2a and 2b) appeared after the thermal reaction (Fig. S1, ESI‡). The isolation of 2a and 2b was achieved using a preparative HPLC system (Fig. S2 and S3, ESI‡). MALDI-TOF mass spectra of 2a and 2b clearly demonstrated the molecular ion peak at m/z 1430 (Fig. S4, ESI‡), and the electron spin resonance (ESR) spectra showed symmetric octet signals in both cases (Fig. S5 and Table S1, ESI‡), confirming that both compounds are pure regioisomers. 1H-NMR signals of anionic forms of 2b (ESR silent), as obtained by bulk electrolysis, revealed the characteristic peaks of the addend (Fig. S6, ESI‡). Indeed, the three singlet signals, at 2.21, 1.27 and 1.19 ppm, are attributed to the methyl protons attached to the pyrrolidine ring, assessing that 2b is a single regioisomer. Although in this work the structure of 2b was not possible to elucidate by X-ray crystallographic analysis, synthetic precedents and theoretical calculations8b strongly suggest that the most feasible addition site of the addend is the one indicated in Scheme 1 (see Fig. S8 (ESI‡) for distinct possible addition patterns). Due to the small amount of 2a obtained from the reaction, NMR characterization and SAM preparation with this compound was not possible to carry out.
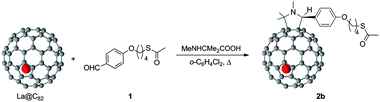 | ||
| Scheme 1 Synthesis of the target derivative of La@C82, as a mixture of regioisomers, from which pure 2b was isolated. | ||
The redox behavior of 2b in ortho-dichlorobenzene (o-DCB) solution (Fig. S9, ESI‡ and Table 1) was studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Clearly reversible peaks on the first reduction and oxidation steps were observed. In addition, it is noteworthy that the first redox potentials of 2b are almost identical to those of pristine La@C82. In other words, 2b maintains the intrinsic electronic properties of the frontier orbitals of pristine La@C82.
| Compound | E ox 2 | E ox 1 | E red 1 | E red 2 |
|---|---|---|---|---|
| a Values are given in volts relative to a Fc0/+ redox couple and were obtained from DPVs. b Conditions: working and counter electrode, Pt wires; reference electrode, Ag/Ag+; supporting electrolyte, 0.1 M TBAPF6 in o-DCB. CV: scan rate, 50 mV s−1. DPV: pulse amplitude, 50 mV; scan rate, 20 mV s−1. c Irreversible. d Data from ref. 10. | ||||
| 2b | 0.48c | 0.05 | −0.35 | −1.58 |
La@C82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
1.07 | 0.07 | −0.42 | −1.34 |
SAMs of 2b on gold (La@C82–S–Au SAM) were formed from a diluted solution of the EMF in o-DCB (67 μM). These conditions were chosen in order to prevent aggregation of the molecules in solution and hence, the consequent deposition of aggregates on the surface. The experimentally optimized conditions for the best quality SAM formation were the following ones: in a glove box filled with nitrogen (humidity around 10%), a freshly cleaned Au(111) on mica was immersed in the EMF solution for at least 48 hours. Then 0.25% in volume of concentrated H2SO4 (98%) was added to deprotect the acetyl group.9 The substrates were removed after 1.5 hours after the acid addition, rinsed with o-DCB and toluene and finally dried under a nitrogen stream.
The modified substrates were characterized by several surface characterization techniques. First, to visualize the surface, Atomic Force Microscopy (AFM) images were acquired (Fig. S10, ESI‡), revealing the formation of a homogenous layer with round-shaped clusters with heights approximately between 0.4 and 1.0 nm. The SAMs were also characterized by high resolution X-Ray Photoelectron Spectroscopy (XPS) (Fig. S11, ESI‡). The La3d spectrum shows the double doublets corresponding to La3d3/2 (856.12 and 850.13 eV) and La3d5/2 (839.00 and 833.69 eV). The S2p spectrum shows a doublet at 161.8 eV and 163 eV assigned to S2p3/2 and S2p1/2 respectively, with a 2:1 intensity ratio and a splitting of 1.2 eV.10 This binding energy is characteristic of the S–Au bond. On the other hand, the absence of a sulfur doublet peak centered at ∼164 eV rules out the presence of surface-unbounded sulfur atoms on the Au surface, that is, the existence of the physisorbed material.9 Additionally, Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) was performed (Fig. S12, ESI‡). The molecular peak is not observed in the spectrum but a peak at m/z 1123 corresponding to the fragment La@C82 is present, which shows the intrinsic isotopic distribution pattern. Consequently, all the above-mentioned techniques clearly elucidated the successful formation of an EMF-based SAM.
As in the case in solution, the electrochemical properties of the resulting SAM were investigated by CV, which revealed that the redox properties of the molecules in solution were also displayed when they are bound to gold. Indeed, two stable and reversible peaks at E1/2 = 0.22 V and 0.57 V (vs. Ag wire) were observed and assigned to the reduction and oxidation of La@C82–S–Au SAM, respectively (Fig. 1). The robustness and stability of the SAM was demonstrated by recording ten consecutive cycles (at 0.3 V s−1), in which no significant loss of current intensity (see Fig. S13a, ESI‡) was found. In addition to that, CVs were carried out at different scan rates (see Fig. S13b, ESI‡) and a linear dependence of the current intensity on the scan rate was observed. This is expected to happen for a rapid reversible redox process of an immobilized redox couple on a surface.11
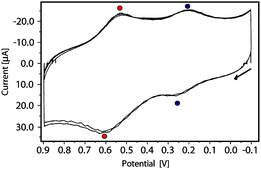 | ||
| Fig. 1 CV obtained by using the La@C822b SAM as a working electrode. NaClO4 20 mM in acetonitrile as electrolyte, with a silver wire and a platinum wire as a pseudo-reference and counter electrode, respectively (scan rate = 0.3 V s−1). In the figure three scans in the range between −0.1 V and 0.9 V are shown. | ||
The magnetic properties of the SAM were investigated by ESR. As expected for a La@C82 derivative which has an open-shell electronic structure, the ESR spectrum of the SAM shows a signal with a g factor of 2.0009 and a linewidth of 4.7 Gauss (Fig. 2a). This result unambiguously demonstrates that the magnetic properties of the EMF are preserved on the surface. The shape of the observed signal is very similar to the one obtained for a drop-cast film (g = 2.0015 and line width of 6.5 Gauss, Fig. S14, ESI‡). These g values are almost identical to those of pristine La@C82 (g = 2.001 and line width ∼10 Gauss) in solids.12 We attribute the low intensity of the ESR signal registered for the SAM to a low spin concentration on the gold surface possibly due to the molecular structure (size and shape) of the grafted molecule that prevents a dense packing. A surface coverage of 1 × 1014 molecules per cm2 has been estimated from the anodic peak of the reduction redox process. This value is lower than the one calculated for other SAMs based on bulky magneto- and electro-active systems.13 This approximate value supports the observed low intensity of the ESR signal. In addition, previously, it was shown for empty fullerenes that the conformational rigidity of the spacer and its physical size play a very important role in their packing. For fullerene derivatives with very similar spacer and anchoring group to the one in 2b, the reported surface area per molecule is much larger than fullerenes with spacers that lead to a less angular shape.14
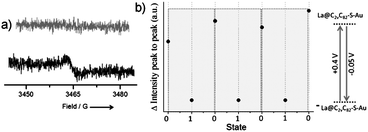 | ||
| Fig. 2 (a) ESR spectra of La@C82–S–Au SAM. Black line: EMF SAM (neutral form); grey in its anionic form. (b) Evolution of the ESR signals (peak to peak intensity) during the potential cycles. One switching cycle (grey boxes) corresponds to the conversion of the SAM between the 0–1–0 states. | ||
Finally, the electrochemical switch between the neutral form (open-shell) and the anionic form (closed-shell electronic structure) of the grafted EMF molecules was monitored by measuring the ESR spectrum after applying a fixed potential to the SAM (Fig. 2). According to the CV, a pulsed potential of −0.05 V was applied (during 3 min) in order to reduce the neutral form of the molecule to the anion, followed by another pulse of +0.4 V (3 min) to promote the reverse redox process. Such sequences of applied pulsed voltages were performed in the electrochemical cell with electrolyte solution. The SAM was then transferred to the ESR cell and measured in air. The oxidized form was not studied in this experiment due to its expected low stability in air.15 In sharp contrast, the one-electron reduced form is considerably stable, as we demonstrated by the isolation of the one-electron reduced anionic form of 2b and its characterization by NMR in solution. As shown in Fig. 2b three consecutive reversible switching cycles (0 → 1 → 0) were performed giving rise to the expected appearance–disappearance of the ESR signal. To support the importance of having covalently bonded molecules and thus, robust SAMs, a drop-cast film based on the pristine non-substituted La@C82 was also prepared and studied. As expected, the electrochemical switch could not be performed due to inefficient electron transport through the thicker film and some desorption of physisorbed molecules.
In summary, a new EMF derivative has been synthesized with the appropriate molecular structure for being covalently grafted on gold. The modified substrates revealed the same electrochemical and magnetic properties of the EMF in solution leading to novel functional surfaces. Finally, the results of the electrochemical switching support the great potential that these materials hold for being used as charge-storage memories devices.
We thank Dr Vega Lloveras for ESR measurements and the support of the Networking Research Center of Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN); the DGI (Spain) (CTQ2010-19501/BQU), the Generalitat de Catalunya (2009SGR00516), and the EU projects ERC StG 2012-306826 e-GAMES and CIG (PCIG10-GA-2011-303989). N.C. thanks the subprogram JdC. This work was also supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas (20108001, “π-Space”), a Grant-in-Aid for Scientific Research (A) (20245006) and (B) (24350019). The iCeMS is supported by World Premier International Research Center Initiative (WPI), MEXT, Japan. We acknowledge CSIC for the publication as Open Access in the RSC.
Notes and references
- (a) M. Rudolf, S. Wolfrum, D. M. Guldi, L. Feng, T. Tsuchiya, T. Akasaka and L. Echegoyen, Chem.–Eur. J., 2012, 18, 5136 CrossRef CAS; (b) Chemistry of Nanocarbons, ed. T. Akasaka, F. Wudl and S. Nagase, Wiley, Chichester, U.K., 2010, p. 275 Search PubMed.
- (a) X. Lu, L. Feng, T. Akasaka and S. Nagas, Chem. Soc. Rev., 2012, 41, 7723 RSC; (b) A. Rodriguez-Fortea, A. L. Balch and J. M. Poblet, Chem. Soc. Rev., 2011, 40, 3551 RSC; (c) S. Fujiki, Y. Kubozono, Y. Rikiishi and T. Urisu, Phys. Rev. B, 2004, 70, 235421 CrossRef.
- (a) M. J. Butcher, J. N. Nolan, M. R. C. Hunt, P. H. Beton, L. Dunsch, P. Kuran, P. Georgi and T. J. S. Dennis, Phys. Rev. B, 2001, 64, 195401 CrossRef; (b) M. J. Butcher, J. W. Nolan, M. R. C. Hunt, P. H. Beton, L. Dunsch, P. Kuran, P. Georgi and T. J. S. Dennis, Phys. Rev. B, 2003, 67, 125413 CrossRef; (c) S. Zhao, J. Zhang, J. Dong, B. Yuan, X. Qiu, S. Yang, J. Hao, H. Zhang, H. Yuan, G. Xing, Y. Zhao and B. Sun, J. Phys. Chem. C, 2011, 115, 6265 CrossRef CAS.
- M. d. C. Gimenez-Lopez, J. A. Gardener, A. Q. Shaw, A. Iwasiewicz-Wabnig, K. Porfyrakis, C. Balmer, G. Dantelle, M. Hadjipanayi, A. Crossley, N. R. Champness, M. R. Castell, G. A. D. Briggs and A. N. Khlobystov, Phys. Chem. Chem. Phys., 2010, 12, 123 RSC.
- M. N. Chaur, F. Melin, A. L. Ortiz and L. Echegoyen, Angew. Chem., Int. Ed., 2009, 48, 7514 CrossRef CAS.
- (a) S. Nagase and K. Kobayashi, J. Chem. Soc., Chem. Commun., 1994, 1837 RSC; (b) D. M. Poirier, M. Knupfer, J. H. Weaver, W. Andreoni, K. Laasonen, M. Parrinello, D. S. Bethune, K. Kikuchi and Y. Achiba, Phys. Rev. B, 1994, 49, 17403 CrossRef.
- (a) S. Sato, S. Seki, Y. Honsho, L. Wang, H. Nikawa, G. Luo, J. Lu, M. Haranaka, T. Tsuchiya, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2011, 133, 2766 CrossRef CAS; (b) T. Tsuchiya, R. Kumashiro, K. Tanigaki, Y. Matsunaga, M. O. Ishitsuka, T. Wakahara, Y. Maeda, Y. Takano, M. Aoyagi, T. Akasaka, M. T. H. Liu, T. Kato, K. Suenaga, J. S. Jeong, S. Iijima, F. Kimura, T. Kimura and S. Nagase, J. Am. Chem. Soc., 2008, 130, 450 CrossRef CAS.
- (a) B. P. Cao, T. Wakahara, Y. Maeda, A. H. Han, T. Akasaka, T. Kato, K. Kobayashi and S. Nagase, Chem.–Eur. J., 2004, 10, 716 CrossRef CAS; (b) Y. Takano, S. Obuchi, N. Mizorogi, R. Garcia, M. Angeles Herranz, M. Rudolf, S. Wolfrum, D. M. Guldi, N. Martin, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 16103 CrossRef CAS.
- Y. Shirai, L. Cheng, B. Chen and J. M. Tour, J. Am. Chem. Soc., 2006, 128, 13479 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083 CrossRef CAS.
- H. X. Ju and D. Leech, Phys. Chem. Chem. Phys., 1999, 1, 1549 RSC.
- R. D. Johnson, M. S. de Vries, J. Salem, D. S. Bethune and C. S. Yannoni, Nature, 1992, 355, 239–240 CrossRef CAS.
- C. Simao, M. Mas-Torrent, J. Veciana and C. Rovira, Nano Lett., 2011, 11, 4382 CrossRef CAS.
- M. d. C. Gimenez-Lopez, M. T. Raeisaenen, T. W. Chamberlain, U. Weber, M. Lebedeva, G. A. Rance, G. A. D. Briggs, D. Pettifor, V. Burlakov, M. Buck and A. N. Khlobystov, Langmuir, 2011, 27, 10977 CrossRef CAS.
- T. Akasaka, T. Wakahara, S. Nagase, K. Kobayashi, M. Waelchli, K. Yamamoto, M. Kondo, S. Shirakura, S. Okubo, Y. Maeda, T. Kato, M. Kako, Y. Nakadaira, R. Nagahata, X. Gao, E. Van Caemelbecke and K. M. Kadish, J. Am. Chem. Soc., 2000, 122, 9316 CrossRef CAS.
Footnotes |
| † Dedicated to Maurizio Prato on his 60th anniversary. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc43578d |
| This journal is © The Royal Society of Chemistry 2013 |