 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sugar-derived organogels as templates for structured, photoluminescent conjugated polymer–inorganic hybrid materials†
Patricia C.
Marr
*a,
Katherine
McBride
a and
Rachel C.
Evans
*b
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, The David Keir Building, Stranmills Road, Belfast, BT9 5AG, UK. E-mail: p.marr@qub.ac.uk; Tel: +44 (0)28 9097 4740
bSchool of Chemistry and CRANN, The University of Dublin, Trinity College, Dublin 2, Ireland. E-mail: raevans@tcd.ie; Tel: +353 1 896 4215
First published on 28th May 2013
Abstract
Co-assembly of an inorganic–organic hybrid material through the combination of supramolecular organogel self-assembly, phase partitioning of a conjugated polymer (CP) and transcription of an inorganic oxide leads to a hybrid material with structured domains of organogel, CP and silica within tube and rod microstructures.
Light-harvesting and light-emitting organic–inorganic hybrids based on conjugated polymers (CPs) show considerable promise for photovoltaic1 and optoelectronic devices.2 Controlled placement of the organic component within the heterogeneous material is challenging due to poor miscibility between the hydrophilic inorganic oxide precursors and the hydrophobic CP. Cooperative physical interactions (e.g. hydrogen bonding, ionic, van de Waals, π–π stacking) at the interface play a crucial role in determining phase miscibility,1a,b,3 morphology3 and the orientation of individual components.4,5 If highly efficient CP–organic–inorganic hybrid devices are to be developed, it is critical that the complex relationship between CP distribution, orientation, interfacial interactions and morphology and macroscopic optoelectronic properties be understood and controlled.1b,6a,b
Strategies explored to address phase separation in CP–organic–inorganic hybrids include in situ polymerisation,7 non-aqueous sol–gel routes,1a,b,2a,3 surfactant templating1a,b,2a,3,5b and the use of water-soluble conjugated polyelectrolytes.6a,c In contrast, phase partitioning has been actively exploited in supramolecular organogels to prepare microstructured inorganic materials.8 Low molecular weight organogelators (LMOGs) are organic molecules which, in an appropriate gelation solvent, can self-assemble into a three-dimensional anisotropic lattice that contains the entrapped solvent.9–13 The resulting matrix can subsequently be employed as a template for the transcription of nanofibrous materials from an inorganic oxide precursor by sol–gel methods.8,14,15 Transcription improves both the thermostability and tensile strength of the gel, or alternatively, removal of the organogel by solvent extraction or sintering, yields the inorganic oxide material, which retains the microstructure of the organic template. Many structures have been obtained, from tubes and rods9–13 to helices8a and double helices.8b In systems where the addition of active species to an organogel/silica transcription medium has been achieved, the additive has been shown to partition with the organogel if it is soluble in the organogel/solvent (e.g. metal complexes) and with silica if it is not soluble (e.g. metal nanoparticles).16 Given the interest in new routes to CP–organic–inorganic hybrids and CP-based nano/microstructures,17 we considered if this approach could be harnessed to prepare CP–inorganic hybrids, in which the relative distribution of the two phases is controlled by phase partitioning.
The sugar-based gelator 1 (Fig. 1) has previously been shown to self-assemble into a fibrous supramolecular organogel network in ethanol.11 Subsequent transcription with tetraethylorthosilicate (TEOS) yielded silica rods and tubes of micron dimensions. Here we show that the addition of a CP as an additive to the organogel mixture, followed by transcription, results in the formation of uniform CP–silica microtubes that retain the photoluminescence (PL) properties of the parent polymer. Phase partitioning of the hydrophobic CP at the organogel–silica interface due to low miscibility with both the polar gelation solvent and the sol–gel precursor leads to spatial segregation of the polymer.
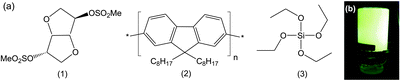 | ||
| Fig. 1 (a) Chemical structures of the gelator 5-di-O-methanesulfonyl-1,4:3,6-dianhydro-D-sorbitol (1), the conjugated polymer PFO (2), and the silica precursor TEOS (3). (b) Inverted PFO-organogel transcribed with silica (PFO-OG-Si) under UV excitation (λex = 366 nm). | ||
Poly(9,9-di-n-octylfluorenyl-2,7-diyl) (PFO) was selected as the CP additive, since its optical properties are known to be highly dependent on the phase morphology.18 For example, the incorporation of PFO into ionic liquid templated silica ionogels leads to cooperative self-assembly of the organic–inorganic components, driving the formation of the PFO β-phase.4 Moreover, poly(fluorenes) can themselves self-assemble into organogels in which supramolecular organisation of the polymer chains in the gel can modify the optoelectronic properties.19
Four samples were investigated: organogel containing PFO (PFO-OG), organogel containing PFO transcribed with silica (PFO-OG-Si) and two blank samples, pure organogel (OG) and organogel transcribed with silica (OG-Si) (for synthetic details see ESI†). The resultant PFO-OG was orange in colour and exhibited yellow-green PL upon excitation at 366 nm (Fig. S1, ESI†). PFO-OG-Si, similarly exhibited yellow-green PL and was stable to inversion (Fig. 1b).
The structural features of the transcribed organogel superstructure were examined using scanning electron microscopy (SEM). Blank transcribed silica tubes (OG-Si) were shown to comprise of both rods and hollow tubes (Fig. 2a), ranging from 10–60 μm in length (mean = 32.5 ± 11.8 μm) and 0.4–1.6 μm (mean = 0.8 ± 0.3 μm) in width, with random orientation. PFO-OG-Si tubes exhibit similar mean dimensions (length = 34.5 ± 26.2 μm, width = 0.8 ± 0.4 μm, aspect ratio ∼ 40) (Fig. 2b); however the sample was more polydisperse and some tubes were able to grow to considerable length without fracture (up to 83.3 μm). The surface morphology of PFO-OG-Si tubes is also smoother (Fig. 2b). Notably, fracturing during preparation of SEM samples was less significant for PFO-OG-Si, suggesting a greater tensile strength than the OG-Si sample. The addition of saturated polymers (e.g. butyl rubber) has previously been shown to enhance the tensile strength of organogels.20
 | ||
| Fig. 2 SEM images and corresponding length distributions of (a) OG-Si and (b) PFO-OG-Si tubes. | ||
29Si MAS NMR indicates the presence of predominantly Q3 ((![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)3SiOH, −102 ppm) and Q4 ((SiO)4Si, −111 ppm) silicon species in PFO-OG-Si (Fig. S5, ESI†). The degree of condensation, C, was determined to be 92%, which is consistent with a highly condensed SiO2 network. Thermogravimetric analysis was used to determine the wt% organic/inorganic content in PFO-OG-Si (Fig. S6, ESI†). The pure organogel OG displays a primary decomposition step corresponding to an 89.9% weight-loss centred at 273 °C, followed by a secondary oxidation process (300–600 °C, 9.2%). PFO undergoes complete (100%) thermal degradation between 300–600 °C.21 On this basis, PFO-OG is estimated to contain ∼78 wt% OG and ∼22 wt% PFO. OG-Si and PFO-OG-Si undergo a complex multistep degradation process, suggesting that the thermal stability of the organic component is improved by the silica tubes. As expected, these samples do not undergo complete thermal degradation under the conditions employed, with the residue at 800 °C being attributed to siliceous species (72 wt% and 56 wt%, respectively). The results indicate that a significant organic component is retained in the sample following filtration, which is confirmed by the FTIR spectrum of PFO-OG-Si (Fig. S7, ESI†). In addition to spectral bands attributed to νSi–O stretching modes (1063, 1166 cm−1), weak bands at 1340 cm−1 and 1757 cm−1, characteristic of OG were also observed. The absence of the νSi–OH stretching mode at 3400 cm−1 also confirms the presence of a condensed siliceous network.
SiO)3SiOH, −102 ppm) and Q4 ((SiO)4Si, −111 ppm) silicon species in PFO-OG-Si (Fig. S5, ESI†). The degree of condensation, C, was determined to be 92%, which is consistent with a highly condensed SiO2 network. Thermogravimetric analysis was used to determine the wt% organic/inorganic content in PFO-OG-Si (Fig. S6, ESI†). The pure organogel OG displays a primary decomposition step corresponding to an 89.9% weight-loss centred at 273 °C, followed by a secondary oxidation process (300–600 °C, 9.2%). PFO undergoes complete (100%) thermal degradation between 300–600 °C.21 On this basis, PFO-OG is estimated to contain ∼78 wt% OG and ∼22 wt% PFO. OG-Si and PFO-OG-Si undergo a complex multistep degradation process, suggesting that the thermal stability of the organic component is improved by the silica tubes. As expected, these samples do not undergo complete thermal degradation under the conditions employed, with the residue at 800 °C being attributed to siliceous species (72 wt% and 56 wt%, respectively). The results indicate that a significant organic component is retained in the sample following filtration, which is confirmed by the FTIR spectrum of PFO-OG-Si (Fig. S7, ESI†). In addition to spectral bands attributed to νSi–O stretching modes (1063, 1166 cm−1), weak bands at 1340 cm−1 and 1757 cm−1, characteristic of OG were also observed. The absence of the νSi–OH stretching mode at 3400 cm−1 also confirms the presence of a condensed siliceous network.
Fig. 3 shows the UV/Vis absorption, PL and excitation spectra for PFO in chloroform and PFO-OG-Si tubes in the solid-state. In CHCl3, the absorption and PL spectra are characteristic of PFO in its disordered α-phase,18 exhibiting a broad absorption band centred at ca. 390 nm and a structured emission band, with peaks at 417, 442 and 471 nm corresponding to 0–0, 0–1 and 0–2 vibronic transitions, respectively. In comparison, the PL spectrum of PFO-OG-Si is red-shifted (439 nm (0–0), 464 nm (0–1) and 493 nm (0–2)) and the relative intensities of the (0–0) and (0–1) vibronic bands are inverted. The corresponding excitation spectrum reveals a broad band between 300–450 nm, with maxima at ∼370 and ∼430 nm. PFO in the β-phase exhibits a characteristic absorption peak at 435 nm and vibrational transitions at 446, 473, and 500 nm in the PL spectrum.4,18 While we cannot exclude the possibility of some β-phase formation, the significant overlap of the excitation/PL spectra, the smaller red-shift in the PL spectrum and the reduced intensity of the 0–0 vibronic band observed for PFO-OG-Si are more indicative of self-absorption. The occurrence of self-absorption implies that the PFO chains are located in close vicinity in the tubes and exist as microscopically segregated PFO domains, as reported previously for poly[9,9-bis(2-ethylhexyl)-fluorene] in saturated polymer blends.22Fig. 3c shows a fluorescence microscopy (FM) image of PFO-OG-Si deposited on a glass substrate. Discrete, well-dispersed microrods are apparent, with uniform PL observed across the full length of the tubes. In contrast, PFO-OG exhibits a heterogeneous dispersion of tube and belt-like structures, with some evidence of bundling (Fig. S8, ESI†). The observed PL appears to be concentrated along the rod/tube edges, which may possibly be due to aggregation of the PFO within a confined spatial region, enhanced PL from emissive surface trap sites, or indeed a combination of both effects.
 | ||
| Fig. 3 (a) Absorption–excitation and (b) PL spectra for PFO in CHCl3 (dashed line) and PFO-OG-Si (solid line) (λex = 370 nm). (c) FM image of PFO-OG-Si (λex = 360–370 nm, 10× magnification). | ||
On the basis of the structural characterisation, we propose that CP-OG-silica microtubes are assembled as follows: the organogelator and solvent are first heated to produce a homogeneous solution. The CP (PFO) solution is added. Upon cooling, self-assembly of the organogel occurs creating a fibrous rod-like structure. Addition of the silica precursor (TEOS) and a suitable catalyst on cooling facilitates transcription (coating of a inorganic sheath onto the surface of an organogel network), which proceeds first by sol deposition, followed by condensation and growth of the silica network. Washing and filtering partially removes the template yielding hollow tubes; where this procedure is insufficient the OG is retained within the core and they appear as rods. PFO is expected to be immiscible with both the organogel and the hydrophilic sol gel phase. Ethanol is employed as the gelation solvent, which, on the basis of their Hildebrand solubility parameters, is expected to be a poor solvent for PFO (δ = 9.1–9.3 and δ = 12.7 for PFO23 and EtOH,24 respectively). PFO is thus expected to exist in an aggregated α-phase (as confirmed by the PL data). 29Si MAS NMR indicates that the silica network is highly condensed, inferring that the PFO is not partitioned within this phase. We propose, that PFO preferentially segregates at the organogel–silica phase interface, (Scheme 1) which is consistent with the observed PL self-absorption and the smooth surface morphology.
 | ||
| Scheme 1 Schematic representation of the proposed structure of a PFO-containing organogel transcribed with silica: self-assembled organic fibre or empty tube (white), PFO (yellow), silica (grey). (a) 3D, (b) cross-section and (c) longitudinal cross section of the hybrid material. | ||
In summary, we have demonstrated the targeted formation of PFO–silica hybrid microtubes using a sugar-derived organogel template, in which the distribution of the organic CP and inorganic silica components was controlled via selective partitioning of the CP at the organogel/silica interface. This mild synthetic approach to CP–organic–inorganic hybrids is expected to be general and extendable to other CPs, enabling the development of CP–inorganic hybrids whose composition and optoelectronic properties are targeted towards an end application. Moreover by changing the gelation solvent it should be possible to modulate the phase partitioning of the CP phase (e.g. from the interface to the organic phase), thereby providing a direct means of controlling the CP distribution within these materials. Mesostructured CP–inorganic oxides have already demonstrated their promise in optoelectronic and photovoltaic device applications.1,2 Moreover one-dimensional CP nanostructures are often touted as potential building blocks for future optoelectronic nanoscale devices.17 Our mild synthetic approach to CP–organic–inorganic hybrid may provide a means of bridging these research fields, leading to the design of more efficient light-emitting and light-harvesting scaffolds.
Solid-state NMR spectra were obtained at the EPSRC UK National Solid-state NMR Service at Durham University.
Notes and references
- (a) S. Neyshtadt, M. Kalina and G. L. Frey, Adv. Mater., 2008, 20, 2541 CrossRef CAS; (b) S. Neyshtadt, J. P. Jahnke, R. J. Messinger, A. Rawal, T. S. Peretz, D. Huppert, B. F. Chmelka and G. L. Frey, J. Am. Chem. Soc., 2011, 133, 10119 CrossRef CAS; (c) T. Xu and Q. Qiao, Energy Environ. Sci., 2011, 4, 2700 RSC; (d) L. Zhao and Z. Lin, Adv. Mater., 2012, 24, 4346 CrossRef CAS.
- (a) E. Dovgolevsky, S. Kirmayer, E. Lakin, Y. Yang, C. J. Brinker and G. L. Frey, J. Mater. Chem., 2008, 18, 423 RSC; (b) A. N. Aleshin, E. L. Alexandrova and I. P Shcherbakov, J. Phys. D: Appl. Phys., 2009, 42, 105108 CrossRef.
- S. Kirmayer, E. Dovgolevsky, M. Kalina, E. Lakin, S. Cadars, J. D. Epping, A. Fernández-Arteaga, C. Rodríguez-Abreu, B. F. Chmelka and G. L. Frey, Chem. Mater., 2008, 20, 3745 CrossRef CAS.
- R. C. Evans and P. C. Marr, Chem. Commun., 2012, 48, 3742 RSC.
- (a) T. Q. Nguyen, J.-J. Wu, B. J. Schwartz and S. H. Tolbert, Science, 2000, 288, 652 CrossRef CAS; (b) Y. Yang, Y. Lu, M. Lu, J. Huang, R. Haddad, G. Xomeritakis, N. Liu, A. P. Malanoski, D. Sturmayr, H. Fan, D. Y. Sasaki, R. A. Assink, J. A. Shellnutt, F. Swol, G. P. Lopez, A. R. Burns and C. J. Brinker, J. Am. Chem. Soc., 2003, 125, 1269 CrossRef CAS; (c) T.-Q. Nguyen, J. Wu, S. H. Tolbert and B. J. Schwartz, Adv. Mater., 2001, 13, 609 CrossRef CAS.
- (a) R. C. Evans, A. G. Macedo, S. Pradhan, U. Scherf, L. D. Carlos and H. D. Burrows, Adv. Mater., 2010, 22, 3032 CrossRef CAS; (b) R. C. Evans, J. Mater. Chem. C, 2013 10.1039/C3TC30543K; (c) S. Clement, A. Tizit, S. Desbief, A. Mehdi, J. D. Winter, P. Gerbaux, R. Lazzaroni and B. Boury, J. Mater. Chem., 2011, 21, 2733 RSC.
- (a) T. Xu, M. Yan, J. D. Hoefelmeyer and Q. Qiao, RSC Adv., 2012, 2, 854 RSC; (b) E. Z. Faraggi, Y. Sorek, O. Levi, Y. Avny, D. Davidov, R. Neumann and R. Reisfeld, Adv. Mater., 1996, 8, 833 CrossRef CAS; (c) B. Luther-Davies, M. Samoc and M. Woodruff, Chem. Mater., 1996, 8, 2586 CrossRef CAS.
- (a) K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980–999 CrossRef CAS; (b) K. Sugiyasu, S.-I. Tamaru, M. Takeuchi, D. Berthier, I. Huc, R. Oda and S. Shinkai, Chem. Commun., 2002, 1212 RSC.
- (a) M. Llusar and C. Sanchez, Chem. Mater., 2008, 20, 782 CrossRef CAS; (b) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS; (c) D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS; (d) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (e) P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699 RSC.
- (a) Molecular Gel Materials with Self-Assembled Fibrillar Networks, ed. P. Terech and R. G. Weiss, Springer, Dordrecht, Netherlands, 2006 Search PubMed; (b) J. W. Steed, Chem. Commun., 2011, 47, 1379 RSC.
- S. J. Craythorne, C. L. Pollock, A. J. Blake, M. Nieuwenhuyzen, A. C. Marr and P. C. Marr, New J. Chem., 2009, 33, 479 RSC.
- A. Vintiloiu and J. C. Leroux, J. Controlled Release, 2008, 125, 179 CrossRef CAS.
- (a) J. Reichwagen, H. Hopf, A. Del Guerzo, C. Belin, J.-P. Desvergne and H. Bouas-Laurent, Org. Lett., 2005, 7, 971 CrossRef CAS; (b) N. M. Sangeetha, S. Bhat, G. Raffy, C. Belin, A. Loppinet-Serani, C. Aymonier, P. Terech, U. Maitra, J. P. Desvergne and A. Del Guerzo, Chem. Mater., 2009, 21, 3424 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol–Gel Science: the Physics and Chemistry of Sol–Gel Processing, Academic Press, 1990 Search PubMed.
- O. Gronwold, E. Snip and S. Shinkai, Curr. Opin. Colloid Interface Sci., 2002, 7, 148 CrossRef.
- A. C. Marr and P. C. Marr, Dalton Trans., 2011, 40, 20 RSC.
- Examples: (a) A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS; (b) J. B. Yu, C. F. Wu, S. P. Sahu, L. P. Fernando, C. Szymanski and J. McNeill, J. Am. Chem. Soc., 2009, 131, 18410 CrossRef CAS; (c) S. Moynihan, D. Iacopino, D. O'Carroll, H. Doyle, D. A. Tanner and G. Redmond, Adv. Mater, 2007, 19, 2474 CrossRef CAS.
- (a) A. P. Monkman, C. Rothe, S. King and F. Dias, Adv. Polym. Sci., 2008, 212, 187 CAS; (b) B. P. Lyons, K. S. Wong and A. P. Monkman, J. Chem. Phys., 2003, 118, 4707 CrossRef CAS.
- (a) J. Lin, Z. Yu, W. Zhu, G. Xing, Z. Lin, S. Yang, L. Xie, C. Niu and W. Huang, Polym. Chem., 2013, 4, 477 RSC; (b) J.-H. Chen, C.-S. Chang, Y.-X. Chang, C.-Y. Chen, H.-L. Chen and S.-A. Chen, Macromolecules, 2009, 42, 1306 CrossRef CAS; (c) Z.-Q. Lin, N.-E. Shi, Y.-B. Li, L. Zhang, J.-Y. Lin, J.-F. Zhao, C. Wang, L.-H. Xie and W. Huang, J. Phys. Chem. C, 2011, 115, 4418 CrossRef CAS.
- D. Ceylan and O. Okay, Macromolecules, 2007, 40, 24 CrossRef.
- C.-H. Chou, S.-L. Hsu, K. Dinakaran, M.-Y. Chiu and K.-H. Wei, Macromolecules, 2005, 38, 745 CrossRef CAS.
- (a) C.-C. Kuo, C.-H. Lin and W.-C. Chen, Macromolecules, 2007, 40, 6959 CrossRef CAS; (b) M. Knaapila, H. L. Vaughan, T. P. A. Hase, R. C. Evans, R. Stepanyan, M. Torkkeli, H. D. Burrows, U. Scherf and A. P. Monkman, Macromolecules, 2010, 43, 299 CrossRef CAS.
- M. Grell, D. D. C. Bradley, X. Long, T. Chamberlain, M. Inbasekaran, E. P. Woo and M. Soliman, Acta Polym., 1998, 49, 439 CrossRef CAS.
- C. E. Carraher Jr, Introduction to Polymer Chemistry, CRC Press, Boca Raton, 3rd edn, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, photographs of materials, SEM images, 29Si MAS-NMR, FTIR spectra, TGA thermograms, surface area, and schematic of transcription process. See DOI: 10.1039/c3cc43320j |
| This journal is © The Royal Society of Chemistry 2013 |