 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Near-IR luminescent neodymium complexes: spectroscopic probes for hydroamination catalysis†
Stacey D.
Bennett
,
Simon J. A.
Pope
and
Benjamin D.
Ward
*
School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: WardBD@Cardiff.ac.uk; Fax: +44 (0)29 208 74030; Tel: +44 (0)29 208 70302
First published on 28th May 2013
Abstract
Neodymium complexes bearing the sensitising bis(oxazolinylphenyl)amine (BOPA) ligands have been prepared, and analysed spectroscopically under both catalytic and pseudo-catalytic conditions with respect to the intramolecular hydroamination of an aminoalkene, providing a direct means of monitoring binding events and relative space around the metal centre.
Rare-earth metal complexes have many applications in catalysis, including hydroamination.1 The presence of partially occupied, low-lying 4f orbitals also gives them unique spectroscopic properties. The formally forbidden f–f transitions are notably sharp compared to many other fluorescence systems, and the long near-IR wavelength provides an emission profile that is largely free from environmentally emissive ‘pollution’.2
The spectroscopic fingerprint of a luminescent lanthanide ion (i.e. spectral profile and lifetime of f-centred emission) is uniquely sensitive to the local coordination environment.3 In particular, the lifetime provides information on the relative shielding of the ion from bound and unbound proximate quenchers, such as O–H, N–H and C–H oscillators,4 present in solvents, (bound) substrates, ligand architectures, and/or the proximity of stereodirecting groups. This parameter may therefore provide reportage on the space at the metal centre in which catalytic reactions take place, and may provide critical information relating to the short-lived species that lie beyond the detection limit of many other analytical techniques.5
Our goal in this study was to use the luminescent properties of Nd(III) to probe the intricate nature of chiral complexes, providing information that cannot be obtained using other spectroscopic techniques, and to relate this information to their behaviour in hydroamination/cyclisation catalysis.
The bis(oxazolinylphenyl)amine (BOPA) series of ligands6 are able to efficiently sensitise the metal-centred excited states of neodymium, such that the resultant near-IR photophysical properties can be used to probe the complexes under conditions relevant to catalytic reactions. In their anionic form, these ligands possess conjugated amides coordinated to the lanthanide in an ideal position to aid investigations in to the luminescence properties of the species.
The protio-ligands react with the neodymium precursor [Nd{N(SiMe3)2}3] to afford the five-coordinate complexes [Nd(R-BOPA){N(SiMe3)2}2] (R = iPr 1a, Ph 1b, Bn 1c) (Scheme 1). The 1H NMR data of the paramagnetic complexes were found to give relatively sharp signals with an expected reduction in fine structure and an increase in chemical shift range (ca. 20 to −40 ppm).7 The overall molecular symmetry, number of resonances, and their relative intensity are fully consistent with the proposed structures. The resonance attributed to the two equivalent N(SiMe3)2 ligands is a distinctive, large singlet in the range of −5.96 to −5.93 ppm. The structure of [Nd(Ph-BOPA){N(SiMe3)2}2] 1b has been confirmed by X-ray crystallography; X-ray data are included in the ESI.†8
![Preparation of [Nd(R-BOPA){N(SiMe3)2}2] 1a–c.](/image/article/2013/CC/c3cc42923g/c3cc42923g-s1.gif) | ||
| Scheme 1 Preparation of [Nd(R-BOPA){N(SiMe3)2}2] 1a–c. | ||
Complexes 1a–c were employed in the catalytic hydroamination of aminoalkenes (vide infra). The catalyst resting state for this reaction (i.e. a tethered amidoalkene) is well understood,9 but cannot be isolated owing to the intramolecular nature of the reaction. Therefore model complexes were prepared that suppress the catalytic turnover by removing the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C from the substrate. The resulting complexes were prepared using the pseudo substrate 2,2′-diphenyl-aminopentane, to afford [Nd(R-BOPA){NHCH2C(Ph2)Pr}2] (R = iPr 2a, Ph 2b, Bn 2c) according to Scheme 2. The 1H NMR data of complexes 3a–c showed similar features to the spectra of 1a–c in terms of the overall C2 symmetry of the complexes. In comparison to the data obtained for complexes 1a–c the signal assigned to the silylamide co-ligands was absent from the spectra of 2a–c.
C from the substrate. The resulting complexes were prepared using the pseudo substrate 2,2′-diphenyl-aminopentane, to afford [Nd(R-BOPA){NHCH2C(Ph2)Pr}2] (R = iPr 2a, Ph 2b, Bn 2c) according to Scheme 2. The 1H NMR data of complexes 3a–c showed similar features to the spectra of 1a–c in terms of the overall C2 symmetry of the complexes. In comparison to the data obtained for complexes 1a–c the signal assigned to the silylamide co-ligands was absent from the spectra of 2a–c.
![Preparation of [Nd(R-BOPA){NHCH2C(Ph)2Pr)}2] 2a–c.](/image/article/2013/CC/c3cc42923g/c3cc42923g-s2.gif) | ||
| Scheme 2 Preparation of [Nd(R-BOPA){NHCH2C(Ph)2Pr)}2] 2a–c. | ||
The spectroscopic analysis of these complexes can only provide adequate information relating to their catalytic performance if supported by a clear demonstration of their implementation in the catalytic reaction being studied. Consequently, complexes 1a–c were employed in the hydroamination/cyclisation of 2,2′-diphenyl-amino-pent-4-ene in toluene-d8 using 10 mol% catalyst at 25 °C (Scheme 3).10 All reactions proceeded to quantitative conversion and with moderate enantioselectivities. The phenyl and benzyl derivatives 1b and 1c gave essentially identical enantiomeric excesses of 44–46%, whereas the isopropyl congener 1a gave a lower level of selectivity (36%). These moderate levels of selectivity can be reasonably explained by the presence of two isomeric forms of the complexes (which the luminescence studies would suggest pertain during the catalytic reaction and that have been identified computationally, vide infra), particularly where one of the components moves the stereodirecting groups away from the expected site of alkene insertion.
![Catalytic hydroamination using [Nd(R-BOPA){N(SiMe3)2}2].](/image/article/2013/CC/c3cc42923g/c3cc42923g-s3.gif) | ||
| Scheme 3 Catalytic hydroamination using [Nd(R-BOPA){N(SiMe3)2}2]. | ||
Solutions of 1a–c (toluene, 10−4 M, N2 atmosphere) were investigated from a photophysical standpoint and are described as dual emissive. Irradiation (λex = 485 nm) induced visible emission from the complexes, typically characterised by a broad, unstructured luminescence band peaking between 500–510 nm with a small associated Stokes' shift (ca. 1000 cm−1).11 The precise positioning of the band and the fluorescence lifetime (5.0, 4.6 and 4.5 ns, for R = iPr, Ph and Bn respectively) were subtly dependent on the specific composition of the ligand. The diphenylamine anion is known to be non-emissive,12 and therefore the visible fluorescence was attributed to a ligand-centred excited state with a contributing charge transfer (N-to-π*) component originating from the deprotonated/coordinated amide bridge-head.
Irradiation of solutions 1a–c at 445 nm also induced sensitised near-IR emission from each of the Nd(III) complexes, with characteristic bands attributed to the 4F3/2–4I9/2, 4F3/2–4I11/2 and 4F3/2–4I13/2 transitions. The corresponding lifetime measurements were also obtained (λem = 1055 nm) and are shown in Table 1; the magnitude of the values are consistent with Nd(III)-centred emission, and each profile fitted reasonably well to a bi-exponential decay yielding two distinct lifetime values. These data suggest that each of the complexes [Nd(R-BOPA){(N(SiMe3)2}2] (1a–c) exists in (at least) two distinct environments. The fact that the analytical and NMR data suggest a single pure complex suggests that these species are likely to be interconverting isomers.
| Compound | Lifetimes (τ)/ns | ||
|---|---|---|---|
| +2,2-Diphenyl-aminopentane | +2,2-Diphenyl-aminopentene | ||
| 1a | 148, 621 (70%) | 111, 267 (59%) (2a) | — |
| 1b | 129, 677 (90%) | 71, 165 (90%) (2b) | — |
| 1c | 41, 308 (80%) | 41, 182 (93%) (2c) | 81, 252 (85%) |
The lifetimes of the two species are remarkably different, and suggest that the main component represents a well-shielded Nd(III) centre, possibly representative of the X-ray structure. The second component (10–30% relative weighting) has a significantly shorter lifetime and suggests a species that is more readily quenched (i.e. by proximate C–H oscillators), possibly arising from a ligand conformation that renders the metal more accessible to solvent molecules. The lifetimes for the isopropyl and phenyl derivatives 1a and 1b are similar, suggesting a comparable environment around the Nd(III) centre, whereas the lifetimes for the benzyl derivative 1c are shorter, implying less effective shielding. This may originate in the greater flexibility of a benzyl (at the methylene group) relative to phenyl and isopropyl groups, which are generally more rigid.
Importantly, the relative intensity of the emission bands of Nd(III) are known to be sensitive to the local coordination environment,3,13 suggesting that changes in either ligand type or ligand arrangement will modulate the emission profile of a luminescent Nd(III) complex. In this context, addition of the saturated pseudo-substrate (2,2′-diphenyl-aminopentane) to form 2a–c, subsequently showed a definitive and measurable change in spectral emission profile (in terms of peak position, profile and integrated intensity, Fig. 1), as well as significant alterations in Nd(III) lifetime. Such observations are clearly indicative of changes in the coordination environment and thus consistent with the bis(trimethylsilyl)amide ligands exchanging with the 2,2′-diphenyl-amino-pentane ligands, establishing that coordination to the Nd(III) must occur via the terminal amino group of the pseudo-substrate. The dominant lifetime components for 2a–c are significantly shortened compared to 1a–c and consistent with a ligand substitution that would introduce N–H oscillators into the coordination sphere and reduce the steric crowding at Nd(III).
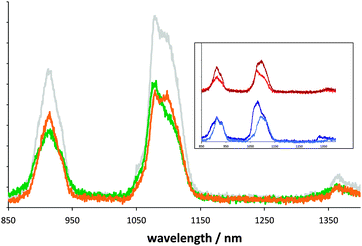 | ||
| Fig. 1 Near-IR luminescence spectra (10−4 M toluene, λex 445 nm). Main: 1c (green), 2c (orange), 1c + 2,2′-diphenyl-amino-pentene (grey). Inset: comparison 1a/2a (light blue/dark blue) and 1b/2b (red/brown). | ||
During the catalytic hydroamination reaction, the proposed cyclised intermediate is coordinated to Nd(III) via a methylene unit.9 Therefore the actual catalytic substrate, 2,2′-diphenyl-amino-pent-4-ene, was also used with 2c and, again, revealed modulation of the emission profile and shortening of the dominant lifetime component to 252 ns. Again, these observations are consistent with a substrate binding event, but one that is distinct from the pseudo-substrate. The lifetime data for this mixture can therefore be differentiated from both the free complex (1c) and the speciation of the saturated substrate analogue (2c).
The identity of the two components of 2a–c (and by extension 1a–c) was probed by calculating the structures of the La(III) congeners; the structures of La-2b are shown in Fig. 2. Calculations suggest that each of the complexes exist in two energetically accessible forms, each with a different sense of helical twist in the diphenyl backbone of the BOPA ligands, giving both exo and endo isomers with respect to the orientation of the stereodirecting groups. The exo isomer is consistent with the X-ray data, and is computationally favoured. It can be seen from Fig. 2 that the metal centre in the endo isomer is significantly less shielded than the exo isomer, and would therefore be more accessible to solvent molecules, accounting for the observed difference in the lifetimes of the two components. Additionally, the relative steric encumbrance of the stereodirecting group is evident, showing that the isopropyl offers the least steric demand and therefore is expected to give less energetic distinction between the isomeric forms, as observed experimentally by luminescence spectroscopy (Table 1).
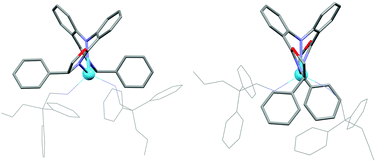 | ||
| Fig. 2 Calculated exo (left) and endo (right) isomers of La-2b. | ||
The exo isomer of a given ligand derivative places the stereodirecting groups in the equatorial plane of the coordination sphere, shielding the metal centre to a greater extent than the endo isomer, which possesses a significant space that is largely unaffected by the ligand periphery (i.e. stereodirecting group). The exo isomer therefore possesses a better-defined chiral space at the coordination sphere, and is more likely to efficiently transfer chiral information to the catalytic substrates. We propose therefore that the relative proportion of these isomers is likely to be a significant contributor to the effectiveness of these complexes to evoke stereoselectivity in catalysis. The experimental data, showing that a higher proportion of the exo isomer is found for the catalyst that generates the highest enantiomeric excess, supports this theory; however this does not contradict the effects of varying the size of the stereodirecting group. Rather, these studies show evidence of additional processes taking place at the molecular level that contribute to the overall effect of controlling stereoselectivity in catalysis.
The photophysics of luminescent neodymium complexes have been used to directly probe the metal species in the catalytic system, with emission profiles before, during, and after giving distinct luminescence signatures. The luminescence studies indicate the presence of different conformations of the complex in solution that are not apparent using NMR spectroscopy and X-ray crystallography. We propose that the presence of these conformations is a key aspect for explaining the relative stereocontrol offered by the different catalyst derivatives.
We thank Cardiff University (Endowment Fellowship to SDB and access to computing facilities “ARCCA”), the Leverhulme Trust, and the EPSRC crystallographic service for support.
Notes and references
- (a) B. D. Ward and L. H. Gade, Chem. Commun., 2012, 48, 10587 RSC; (b) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS; (c) G. A. Molander and J. A. C. Romero, Chem. Rev., 2002, 102, 2161 CrossRef CAS; (d) Z. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1 CrossRef CAS.
- J.-C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939 RSC.
- (a) J.-C. G. Bünzli, A.-S. Chauvin, H. K. Kim, E. Dieters and S. V. Eliseeva, Coord. Chem. Rev., 2010, 254, 2623 CrossRef; (b) S. Faulkner, S. J. A. Pope and B. P. Burton-Pye, Appl. Spectrosc. Rev., 2005, 40, 1 CrossRef CAS.
- (a) W. D. Horrocks Jr. and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384 CrossRef; (b) E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542 CrossRef CAS; (c) A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- (a) X. Zhu, W.-K. Wong, W.-Y. Wong and X. Yang, Eur. J. Inorg. Chem., 2011, 4651 CrossRef CAS; (b) B. K. McMahon and T. Gunnlaugsson, J. Am. Chem. Soc., 2012, 134, 10725 CrossRef CAS; (c) W. Chen, B. D. Wright and Y. Pang, Chem. Commun., 2012, 48, 3824 RSC.
- (a) H. A. McManus and P. J. Guiry, J. Org. Chem., 2002, 67, 8566 CrossRef CAS; (b) S.-F. Lu, D.-M. Du, S.-W. Zhang and J. Xu, Tetrahedron: Asymmetry, 2004, 15, 3433 CrossRef CAS; (c) H. A. McManus, P. G. Cozzi and P. J. Guiry, Adv. Synth. Catal., 2006, 348, 551 CrossRef CAS; (d) T. Inagaki, L. T. Phong, A. Furuta, J.-I. Ito and H. Nishiyama, Chem.–Eur. J., 2010, 16, 3090 CrossRef CAS.
- (a) L. Lukešová, B. D. Ward, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Dalton Trans., 2007, 920 Search PubMed; (b) L. Lukešová, B. D. Ward, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Organometallics, 2007, 26, 4652 CrossRef.
- X-ray data were collected by the EPSRC National Crystallographic Service: S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- (a) M. R. Gagnè, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275 CrossRef; (b) Y. Li and T. J. Marks, J. Am. Chem. Soc., 1996, 118, 9295 CrossRef; (c) A. Motta, G. Lanza, I. L. Fragalà and T. J. Marks, Organometallics, 2004, 23, 4097 CrossRef CAS.
- For selected examples of asymmetric hydroamination catalysis using lanthanides see: (a) Y. Li and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef; (b) S. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 14768 CrossRef CAS; (c) P. N. O'Shaughnessy and P. Scott, Tetrahedron: Asymmetry, 2003, 14, 1979 CrossRef CAS; (d) P. N. O'Shaughnessy, P. D. Knight, C. Morton, K. M. Gillespie and P. Scott, Chem. Commun., 2003, 1770 RSC; (e) P. N. O'Shaughnessy, K. M. Gillespie, P. D. Knight, I. J. Munslow and P. Scott, Dalton Trans., 2004, 2251 RSC.
- For a comprehensive and authoritative review of issues relating to fluorescence see J. R. Lakowitz, Principles of Fluorescence Spectroscopy, Springer, New York, third edn, 2006 Search PubMed.
- N. Chattopadhyay, A. Samanta, T. Kundu and M. Chowdhury, J. Photochem. Photobiol., A, 1989, 48, 61 CrossRef CAS.
- S. J. A. Pope, B. P. Burton-Pye, R. Berridge, T. Khan, P. J. Skabara and S. Faulkner, Dalton Trans., 2006, 2907 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, crystal, computational and characterising data for all new complexes. CCDC 903609. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc42923g |
| This journal is © The Royal Society of Chemistry 2013 |