 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescence-activated cell sorting and directed evolution of α-N-acetylgalactosaminidases using a quenched activity-based probe (qABP)†
Kamaladasan
Kalidasan‡
a,
Ying
Su‡
b,
Xiaoyuan
Wu‡
b,
Shao Q.
Yao
*b and
Mahesh
Uttamchandani
*abc
aDefence Medical and Environmental Research Institute, DSO National Laboratories, 27 Medical Drive, Singapore 117510, Singapore
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmyaosq@nus.edu.sg; Fax: +65 6779 1691; Tel: +65 6516 2925
cDepartment of Biological Science, National University of Singapore, 14 Science Drive 4, Singapore 117543, Singapore. E-mail: mahesh@dso.org.sg; Fax: +65 6485 7033; Tel: +65 6485 7214
First published on 19th June 2013
Abstract
An α-N-acetylgalactosamine-containing quenched activity-based probe (qABP) was designed and successfully synthesized, and it was subsequently used in directed enzyme evolution experiments aided by fluorescence-activated cell sorting (FACS) for the discovery of an α-N-acetylgalactosaminidase variant with improved catalytic activity.
Fluorescence-activated cell sorting (FACS), when combined with random mutagenesis, offers a potent strategy for directed protein evolution.1–3 At the heart of this technique are the range of proteins, dyes or probes which transduce the intended biological event into quantifiable fluorescent signals in cells.4,5 Both fluorogenic substrates and activity-based probes (ABPs) may conceivably be used to drive selection using FACS, enabling potential high-throughput identification and separation of cell populations, which express desirably active protein variants.6,7 Current strategies are however caught between two extremes: fluorogenic probes upon activation easily diffuse out of cells, diminishing the positive fluorescent readout needed for FACS selection.8,9 Conversely, ABPs designed as suicide inhibitors are persistently fluorescent (always on, resulting in higher backgrounds/false positives) and bind to the enzyme active site covalently, limiting the catalytic turnover and positive readout.4,10
In order to address these limitations, we herein report the design of a ‘smart’ quinone methide-based quenched activity-based probe (qABP) with the following special features: (1) a modular structure, (2) turn-on fluorescence, (3) ability to diffuse away from the enzyme active site (facilitating multiple turnovers from a single enzyme molecule) and (4) retention within the cell (thus magnifying fluorescent signals in target cell populations). We have previously applied our probe design to image target enzymes, specifically caspases and phosphatases, within precise sub-cellular locations of mammalian cells.11 While Kwan et al. have similarly applied quinone methide-based probes to isolate cells expressing or not expressing certain glycosidases,12 we demonstrate here, for the first time, the utility of such probes for evolving galactosidase activities, using error-prone PCR and rounds of FACS selection of mutant libraries, in E. coli (Fig. 1A).
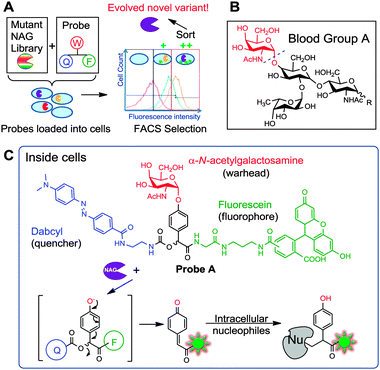 | ||
| Fig. 1 (A) Overall strategy of evolving enzymes using a smart qABP and FACS. The mutant library is sorted and subjected to FACS. (B) Structure of glycolipids on the surface of Type A red blood cells, which upon cleavage by NAG at the position indicated with a dotted line would produce Type O RBCs. (C) Structure of Probe A and mechanism of action. W: warhead; F: fluorophore; Q: quencher. | ||
We elected to apply this strategy to a unique α-N-acetyl-galactosaminidase (NAG), recently discovered from a soil bacterium Elizabethkingea miricola.13 This enzyme was found to selectively hydrolyse the α-N-acetyl-galactosamine (A-antigen) of red blood cells (RBCs) surface glycosides, with potential applications in converting “A” type blood stocks to the universal “O” type blood (Fig. 1B).14,15 At current activities, sixty milligrams of the wildtype NAG enzyme is required to convert one pint of packed RBCs (200 ml volume) under a 1 h incubation.13 At these quantities, scaling up can be prohibitively costly, hence there is a need for more efficient enzymes.
Probe A was designed with an α-N-acetyl-galactosamine warhead (W), a fluorescence reporter fluorescein (F) and a dabcyl quencher (Q), built around a mandelic acid core (Fig. 1C). Upon intracellular cleavage by NAG to release the warhead, a 1,6-elimination reaction would liberate the dabcyl module, generating fluorescence and a reactive quinone methide intermediate that would subsequently covalently react with nearby intracellular nucleophiles (Fig. 1C). Such probes are ill-suited as ABPs for target identification as they diffuse away from the enzyme active site. Nevertheless, for bioimaging of localized intracellular enzyme activities as previously demonstrated,11 as well as directed evolution using FACS, which is reported in the current study, this is exactly the property that we intended to exploit, to retain enzymatic activity while magnifying differences across variants.
Probe A was synthesized over 11 solution-phase steps (Scheme 1 and Scheme S1, ESI†). Briefly, the commercial galactosamine 1 was converted to the corresponding azide and reacted with acetic anhydride, giving 2. The hydroxide at position 1 was selectively deprotected, giving 3. Upon introduction of the mandelic acid core, the corresponding azide intermediate 5 was obtained. Direct conversion of N3 in 5 to NHAc was achieved in a single-step, giving 6. Next, dabcyl-NH2 was introduced, producing 8. Finally, fluorescein was introduced to produce 10, which upon further deprotection afforded Probe A.
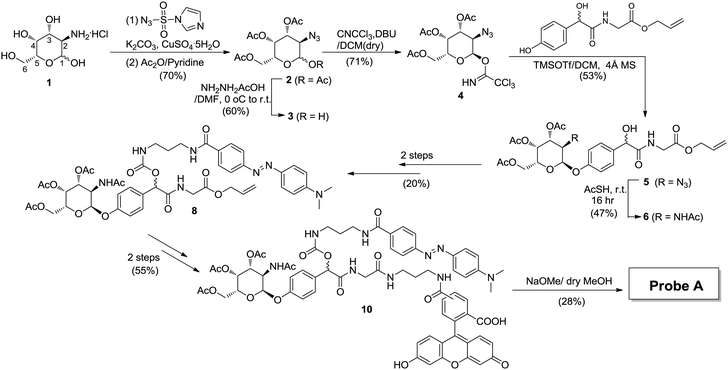 | ||
| Scheme 1 Probe synthesis strategy. Fluorescein was selected as the fluorophore and dabcyl as the quencher. | ||
Next, NAG was cloned into pDEST17 using the Gateway cloning system (Invitrogen) (Table S1, ESI†). The library was generated using error-prone PCR (see ESI†) which was then transformed into NovaBlue(DE3) competent cells by electroporation. These cells were well suited as their LacZ (β-galactosidase) gene was disrupted. They also possessed a T7 promoter system compatible with pDEST17. The purified wildtype NAG enzyme was well-expressed and active (Fig. 2).
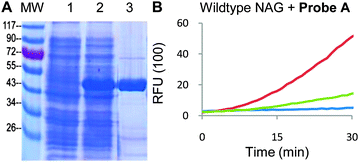 | ||
| Fig. 2 (A) Purified NAG derived from Elizabethkingia miricola. Lanes – MW: molecular weight marker (sizes in kD); (1) uninduced lysates; (2) induced lysates; (3) purified NAG protein (expected MW 46 kD). (B) Concentration-dependent activity of WT NAG against 20 μM Probe Ain vitro. Red curve − 20 nM NAG; green curve − 2 nM NAG; blue curve – blank, with probe only. Readouts obtained at λ490 nm/520 nm. | ||
We initially tested the purified NAG enzyme with commercial coumarin Aα probes in vitro (Fig. S1A, ESI†). As expected, these probes provided high fluorescent readouts, confirming the expected enzymatic activity (Fig. S1B, ESI†). When tested with Probe A, pure NAG also exhibited strong enzyme activity-dependent fluorescent readouts, establishing compatibility with our probes in vitro (Fig. 2B and Fig. S1C, ESI†). NAG-expressing cell lysates also produced positive readouts (Fig. S1, ESI†). The probes were also tested against the NovaBlue(DE3) lysates, confirming no background activity. By running the probe-labeled lysates on the gel followed by in-gel fluorescence scanning, it was revealed that a broad spectrum of endogenous proteins was labeled by Probe A in the presence of active NAG (Fig. S2, ESI†), indicating successful intracellular trapping of the probe.
Probe delivery into E. coli cells was vital for cell-based screening of enzyme activity during FACS analysis, and we needed to establish an effective cell-loading protocol for the probe. The performance of loading was measured using FACS readouts with NAG-overexpressing NovaBlue(DE3) cells (Fig. 3A). Upon optimization (see ESI†), we were able to find conditions under which a normally distributed population of positively fluorescent cells was obtained consistently. A significant proportion of the cell population generated fluorescence readouts above 100 RFUs, with greater than 95% of the cell population producing consistent readouts greater than 10 RFUs. By incubating the cell population for extended period of times on ice (to limit cell division), we found that the cells produced nearly identical sorting profiles, and the probe was successfully retained in the cells for at least several hours without significant loss of fluorescence.
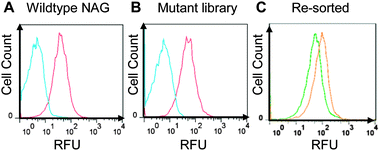 | ||
| Fig. 3 FACS sorting results. (A) Wildtype NAG expressing cells sorted with and without Probe A (red and blue histograms respectively). (B) Mutant library sorted with and without probe (red and blue histograms respectively). (C) The bottom 0.5% and the top 0.2% library populations were re-grown and sorted, generating the distinct green and orange histograms respectively. | ||
Thereafter, we tested the library of NAG variants with Probe A. We found that a large fraction of electroporated mutant library cells incubated with Probe A emitted fluorescence (Fig. 3B, red curve), when compared to the group that contained Novablue(DE3) cells without Probe A (Fig. 3B, blue curve). In order to test that we could select desired populations of active variants, we proceeded to sort out the top 0.2% of the electroporated mutant library. We also sorted the bottom 0.5% of the library. These two populations were then re-grown in culture and studied using FACS (Fig. 3B, orange and green curves respectively). The results showed that there was a marked difference in the activity of the sorted cell populations, indicating that higher-activity variants could be enriched using the strategy. We thus proceeded to select novel NAG variants with enhanced activity. A further top 0.2% of cells were sorted in the second round. After each round, half of the sorted samples were plated while the other half were grown for another round of sorting. Individual colonies were randomly sequenced colonies from each round of sorting (representative results in Table S3, ESI†).
The NAG enzyme is grouped within the GH109 family of glycosidases. Its structure has been established, revealing residues Tyr-307, Tyr-225, His-228 and Glu-149 are involved with substrate binding in the enzyme active site.13 Based on the sequences and structural positions of mutation sequences, we selected five Round-2 colonies with mutations near the active site for follow-up (Table S3, ESI†). However, we could only successfully purify the enzyme from one of the colonies and hence quantified its activity profiles carefully. The yields from the other variants were extremely low, when purification was attempted under native conditions. The purified variant carried two non-synonymous mutations (H104L and S347R) (Table S2 and Fig. S3, ESI†). Both these mutations are located within 20 Å of the enzyme active site (Fig. 4C). This variant NAG was found to exhibit a lower KM and close to two times higher kcat/KM ratio than the wildtype NAG (Fig. 4). This correlated well with expectations from the FACS experiments. No further variants with enhanced activities were identified.
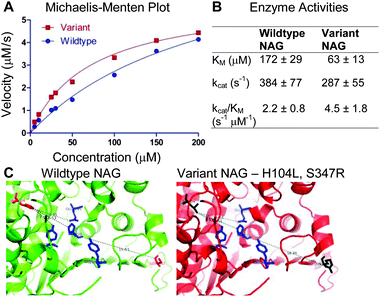 | ||
| Fig. 4 (A) Kinetic activities of the control and the variant NAG enzymes (H104L, S347R) were plotted on a Michaelis–Menten plot (averaged from triplicates). (B) KM and kcat values of the NAG wildtype and variant were calculated by non-linear regression, least squares fitting, ±95% confidence intervals. (C) The wildtype NAG enzyme is shown on the left. The variant NAG enzyme (with protein ribbon in red) has mutated amino acids H104L, S347R in black. The active site residues are shown in blue. Figures are adapted from PDB entry 2IXA13 and rendered in Pymol. | ||
In conclusion, we evolved a sub-optimal enzyme to a more active form using the smart qABP strategy developed herein. Compared to traditional fluorogenic probes, quinone methide qABPs do not diffuse out of cells readily, so continued measurements of the fluorescence signals will be possible. As far as we know, this is the first example of using such smart quinone methide-based probes for evolving enzyme activity. The novel NAG variant identified will be tested on human RBCs to evaluate its efficacy. We anticipate that this strategy may be applied more broadly in enzyme engineering and evolution.
The authors acknowledge funding support from DSO National Laboratories, Singapore (Grant no. 20090211). Ms Tan Pei Xin developed the NAG constructs used in this study.
Notes and references
- G. Yang and S. G. Withers, ChemBioChem, 2009, 10, 2704–2715 CrossRef CAS.
- R. E. Cobb, T. Si and H. Zhao, Curr. Opin. Chem. Biol., 2012, 16, 285–291 CrossRef CAS.
- M. Goldsmith and D. S. Tawfik, Curr. Opin. Struct. Biol., 2012, 22, 406–412 CrossRef CAS.
- M. Uttamchandani, J. Li, H. Sun and S. Q. Yao, ChemBioChem, 2008, 9, 667–675 CrossRef CAS.
- Y. Urano, Curr. Opin. Chem. Biol., 2012, 16, 602–608 CrossRef CAS.
- G. Yang, J. R. Rich, M. Gilbert, W. W. Wakarchuk, Y. Feng and S. G. Withers, J. Am. Chem. Soc., 2010, 132, 10570–10577 CrossRef CAS.
- S. Bershtein and D. S. Tawfik, Curr. Opin. Chem. Biol., 2008, 12, 151–158 CrossRef CAS.
- R. P. Haugland, Biotech. Histochem., 1995, 70, 243–251 CrossRef CAS.
- B. Z. Packard and A. Komoriya, Cell Res., 2008, 18, 238–247 CrossRef CAS.
- G. Blum, S. R. Mullins, K. Keren, M. Fonovic, C. Jedeszko, M. J. Rice, B. F. Sloane and M. Bogyo, Nat. Chem. Biol., 2005, 1, 203–209 CrossRef CAS.
- M. Hu, L. Li, H. Wu, Y. Su, P. Y. Yang, M. Uttamchandani, Q. H. Xu and S. Q. Yao, J. Am. Chem. Soc., 2011, 133, 12009–12020 CrossRef CAS.
- D. H. Kwan, H. M. Chen, K. Ratananikom, S. M. Hancock, Y. Watanabe, P. T. Kongsaeree, A. L. Samuels and S. G. Withers, Angew. Chem., Int. Ed., 2011, 50, 300–303 CrossRef CAS.
- Q. P. Liu, G. Sulzenbacher, H. Yuan, E. P. Bennett, G. Pietz, K. Saunders, J. Spence, E. Nudelman, S. B. Levery, T. White, J. M. Neveu, W. S. Lane, Y. Bourne, M. L. Olsson, B. Henrissat and H. Clausen, Nat. Biotechnol., 2007, 25, 454–464 CrossRef CAS.
- J. Goldstein, G. Siviglia, R. Hurst, L. Lenny and L. Reich, Science, 1982, 215, 168–170 CrossRef CAS.
- L. L. Lenny, R. Hurst, J. Goldstein, L. J. Benjamin and R. L. Jones, Blood, 1991, 77, 1383–1388 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis scheme and characterization of intermediates, microplate/gel results and FACS optimization. See DOI: 10.1039/c3cc42836b |
| ‡ Joint first authors. |
| This journal is © The Royal Society of Chemistry 2013 |