 Open Access Article
Open Access ArticleDibenzyl and diallyl 2,6-bisiminopyridinezinc(II) complexes: selective alkyl migration to the pyridine ring leads to remarkably stable dihydropyridinates†
John J.
Sandoval
,
Pilar
Palma
,
Eleuterio
Álvarez
,
Antonio
Rodríguez-Delgado
* and
Juan
Cámpora
*
Instituto de Investigaciones Químicas, CSIC - Universidad de Sevilla, c/Americo Vespucio 49, 41092, Sevilla, Spain. E-mail: campora@.iiq.csic.es; arodriguez@us.es; Fax: +34 954460565; Tel: +34 954489555
First published on 7th June 2013
Abstract
Diorganozinc compounds (ZnR2) with R = CH2Ph or CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 react with 2,6-bisiminopyridines (iPrBIP) to afford thermally stable dihydropyridinate(−1) complexes, and do not react if R = CH2SiMe3 or CH2CMe2Ph. NMR studies reveal that dibenzylzinc binds iPrBIP at −80 °C, yielding the unstable complex [Zn(CH2Ph)2(iPrBIP)]. Above −20 °C, this undergoes selective alkyl migration to the remote 4 position of the central pyridine ring.
CH2 react with 2,6-bisiminopyridines (iPrBIP) to afford thermally stable dihydropyridinate(−1) complexes, and do not react if R = CH2SiMe3 or CH2CMe2Ph. NMR studies reveal that dibenzylzinc binds iPrBIP at −80 °C, yielding the unstable complex [Zn(CH2Ph)2(iPrBIP)]. Above −20 °C, this undergoes selective alkyl migration to the remote 4 position of the central pyridine ring.
Bisiminopyridines (BIP) are emerging as promising ligands in catalysis, not only in olefin polymerization or oligomerization,1 but also in other important transformations such as hydrogenation,2 hydrosilylation,3 cross-coupling reactions4 or alkene hydrocarboxylation.5 Their success is linked to their non-innocent behaviour that imparts rather unique redox properties to their complexes.6,7 Another consequence of this redox non-innocence is the extensive and often unpredictable ligand-centered reactivity of BIP complexes that frequently leads to novel molecular transformations. For example, the reaction of diorganomanganese compounds with BIP ligands involves selective alkyl transfer from the Mn(II) center to the 4 position of the central pyridine.8 We exploited this reactivity to develop a simple and quite general methodology for the synthesis of functionalized BIP ligands9 that can later be used for different purposes.10
One of the main difficulties associated with high-valent organomanganese compounds is that they are usually paramagnetic and NMR silent, precluding direct investigation of their reactivity by NMR techniques. On the other hand, the common high-spin configuration of Mn(II), with all five d orbitals half-filled, results in a chemical behaviour more similar to main group elements than to transition metals.11 This led us to wonder if diamagnetic dialkylzinc(II) compounds (with closed shell d10 configuration) would react with BIP ligands in the same way as their manganese analogues. Some years ago, Gibson studied the reactions of dimethyl or diethylzinc with BIP ligands. Surprisingly, ZnMe2 is unreactive, but ZnEt2 gives rise to different products resulting from alkyl migration to the ligand, either to the N atom or to the carbon at position 2, depending on the steric bulk of the side N-aryl substituents of the ligand.12 These results contrast with our Mn(II) chemistry, but since Gibson's work was limited to the simplest ZnR2 derivatives (R = Me, Et), we decided to investigate similar reactions with R = CH2SiMe3, CH2CMe2Ph, CH2Ph and CH2CHCH2, which we had previously used in our work with Mn(II) complexes. Herein, we describe the results of this study. We also provide some insights into the mechanism of the alkyl transfer reaction, including direct detection of an unstable dialkyl bisiminopyridine complex and its quantitative conversion into the bisiminodihydropyridinate(−1) derivative.
Treatment of iPrBIP with ZnR2 generated in situ by the reaction of the corresponding Grignard reagents with ZnCl2 in THF led to different results depending on the chosen alkyl (Scheme 1). When iPrBIP is treated at −80 °C with stoichiometric amounts of solutions of diallylzinc or dibenzylzinc, striking colour changes happen. As the mixtures warm, their colours evolve towards dark purple-blue hues that remain at room temperature. Quenching these mixtures with anhydrous methanol followed by extraction in hexane and filtration affords high yields of known9 2,6-diimino-4-alkyl-1,4-dihydropyridines La and Lb, containing only small amounts (ca. 5%) of the corresponding aromatized pyridines La′ and Lb′. In contrast, no colour changes were observed when iPrBIP was similarly mixed with bis(trimethylsilymethyl)zinc or dineophylzinc and the resulting mixtures were stirred for several hours at room temperature, or heated to 90 °C. Treatment with methanol, followed by the above-described workup led to the recovery of unaltered iPrBIP.
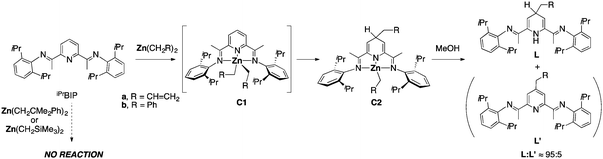 | ||
| Scheme 1 | ||
Our results are in accordance with Gibson's findings in the sense that the reactivity of zinc dialkyls with BIP ligands is strongly dependent on the nature of the R group. As Gibson points out, differences in the regioselectivity of alkyl migration are probably due to steric effects. Nevertheless, we have also shown that dibenzyl or diallylzinc behave similarly to their Mn(II) analogues. The manganese and zinc-based reactions however differ in the different ratios of dihydropyridine (La, Lb) to aromatized pyridine (La′, Lb′) products. In the Mn(II) system, aromatized products result from the slow but spontaneous dehydrogenation of the Mn analogue of C2 to give a 4-alkylpyridine complex.9,10a Since the pyridine/dihydropyridine ratio depends on the rate of the dehydrogenation process and prescribed reaction time, aromatized pyridines can become main products. In contrast, the fractions of La′ or Lb′ obtained with zinc reagents do not increase when the reaction time is extended up to 24 h. This led us to suspect that the small amount of La′ and Lb′ obtained with zinc reagents are not due to spontaneous dehydrogenation of dihydropyridinate intermediates, but due to the oxidation of free ligands La and Lb during the reaction workup. In order to confirm this point, we resolved to isolate and characterize the dihydropyridinate complexes, C2. Omitting the methanol quenching step and after adequate workup to remove salts and excess of other reagents, purple crystals of the benzyl derivative C2b were obtained. As expected, the allylzinc derivative C2a proved to be very reactive and sensitive to traces of oxygen, which prevented us from isolating it in pure form, although it could be unambiguously identified in the 1H NMR spectra of crude samples (see ESI†). Apart from their reactivity, complexes C2 are thermally stable, and remain in solution at room temperature for extended periods. Monitoring a C6D6 solution of C2b in a Teflon-valve screw cap sealed NMR tube for 15 days showed no signs of decomposition, decaying less than 10% when heated at 90 °C for 24 h.
Although a number of complexes resulting from alkyl transfer from the metal to the BIP framework have been reported,10c,12 the crystal structure of C2b (Fig. 1) is the first for a BIP complex that exhibits alkylation of the C4 atom of the central heterocyclic ring. The zinc center is in a distorted square planar coordination environment. The distances from zinc to the imine nitrogen atoms, 2.3006(16) and 2.3545(17) Å, are normal for coordinate bonds, whereas the bond to the central N atom is considerably shorter, 1.9093(16) Å. Bonds in the central 1,4-dihydropyridine ring show alternating lengths as expected for the two localized CC bonds (e.g., C1–C2, 1.350(3); C2–C3, 1.499(3) Å).
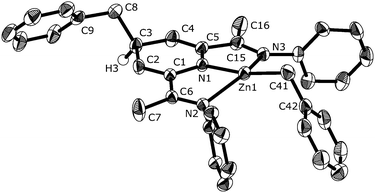 | ||
| Fig. 1 ORTEP plot of C2b showing ellipsoids drawn at 50% probability level. i-Pr groups have been omitted for clarity. | ||
As shown in Scheme 1, complexes C2 must originate from C1, which undergoes alkyl migration. In order to gain further understanding of the mechanism of this transformation, we prepared samples of pure dialkyls ZnR2 (R = CH2SiMe3, CH2CMe2Ph and CH2Ph), and studied their reactions with iPrBIP using NMR. Diallylzinc was not included due to its low thermal stability.13 As expected, no reaction took place when iPrBIP was treated with Zn(CH2SiMe3)2 or Zn(CH2CMe2Ph) in C6D6 or CD2Cl2. No interaction was detected in the 1H NMR spectra of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of Zn(CH2SiMe3)2 and iPrBIP between −80 and +90 °C. In contrast, iPrBIP reacts with dibenzylzinc instantaneously at 23 °C, affording C2b. A rapid deep colour change was also observed when the reagents were combined in CD2Cl2 at −80 °C. The 1H NMR spectrum of this mixture shows quantitative transformation into a new species, C1b. Fig. 2 shows the evolution of this compound as the temperature is gradually increased. Above −20 °C, signals corresponding to C2b grow at the expense of those of C1b, which fade off when the solution reaches room temperature. Accordingly, compound C2b was obtained in high yield when this reaction was repeated at a preparative scale. Metal dialkyl complexes [MR2(BIP)] have proven to be elusive species,9,12f,14 as they are usually unstable unless the alkyl groups are stabilized with β-silyl groups (e.g., R = CH2SiMe3). C1b is the first ever detected complex of this class containing normal alkyl groups. The 1H NMR spectrum of C1b, recorded at −70 °C, is fully consistent with that of a bis(benzyl)zinc–BIP complex. Two signals in the low field region of the 1H NMR spectrum of this compound, a triplet (δ 8.00) and a doublet (δ 7.55) with a 2:1 intensity ratio indicate that the pyridine ring retains its aromaticity. Both Zn-bound CH2 groups give rise to a single resonance for 4H at δ 1.22, but five signals are observed for the benzyl Ph protons in the 6.5 and 5.5 ppm region, indicating the chemical inequivalence of the benzyl groups: two doublets for ortho, two triplets for meta and one multiplet for the overlapping resonances of the para phenyl hydrogen atoms. Thus, the metal center is probably in a square pyramidal environment, with one benzyl group at the apex, and the other at the base. This configuration breaks the symmetry of the N-aryl substituents of the BIP ligands, splitting the i-Pr group signals. As seen in Fig. 2, warming to −30 °C causes the simplification of the spectrum that shows single sets of signals for the benzyl and the N-aryl groups. The fluxional behaviour evidences rapid swinging of the benzyl groups between the apical and basal positions. Coincident values of 10.5 kcal mol−1 are independently obtained for the energy barrier from the coalescence temperatures of the signals of the o-Ph protons and the methyne groups of the i-Pr substituents. This is essentially the same value reported for the swinging of the alkyl groups in the related iron complex [Fe(CH2SiMe3)2(iPrBIP)] (ca. 10 kcal mol−1).14b
:1 mixtures of Zn(CH2SiMe3)2 and iPrBIP between −80 and +90 °C. In contrast, iPrBIP reacts with dibenzylzinc instantaneously at 23 °C, affording C2b. A rapid deep colour change was also observed when the reagents were combined in CD2Cl2 at −80 °C. The 1H NMR spectrum of this mixture shows quantitative transformation into a new species, C1b. Fig. 2 shows the evolution of this compound as the temperature is gradually increased. Above −20 °C, signals corresponding to C2b grow at the expense of those of C1b, which fade off when the solution reaches room temperature. Accordingly, compound C2b was obtained in high yield when this reaction was repeated at a preparative scale. Metal dialkyl complexes [MR2(BIP)] have proven to be elusive species,9,12f,14 as they are usually unstable unless the alkyl groups are stabilized with β-silyl groups (e.g., R = CH2SiMe3). C1b is the first ever detected complex of this class containing normal alkyl groups. The 1H NMR spectrum of C1b, recorded at −70 °C, is fully consistent with that of a bis(benzyl)zinc–BIP complex. Two signals in the low field region of the 1H NMR spectrum of this compound, a triplet (δ 8.00) and a doublet (δ 7.55) with a 2:1 intensity ratio indicate that the pyridine ring retains its aromaticity. Both Zn-bound CH2 groups give rise to a single resonance for 4H at δ 1.22, but five signals are observed for the benzyl Ph protons in the 6.5 and 5.5 ppm region, indicating the chemical inequivalence of the benzyl groups: two doublets for ortho, two triplets for meta and one multiplet for the overlapping resonances of the para phenyl hydrogen atoms. Thus, the metal center is probably in a square pyramidal environment, with one benzyl group at the apex, and the other at the base. This configuration breaks the symmetry of the N-aryl substituents of the BIP ligands, splitting the i-Pr group signals. As seen in Fig. 2, warming to −30 °C causes the simplification of the spectrum that shows single sets of signals for the benzyl and the N-aryl groups. The fluxional behaviour evidences rapid swinging of the benzyl groups between the apical and basal positions. Coincident values of 10.5 kcal mol−1 are independently obtained for the energy barrier from the coalescence temperatures of the signals of the o-Ph protons and the methyne groups of the i-Pr substituents. This is essentially the same value reported for the swinging of the alkyl groups in the related iron complex [Fe(CH2SiMe3)2(iPrBIP)] (ca. 10 kcal mol−1).14b
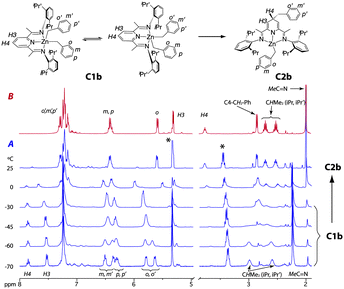 | ||
| Fig. 2 (A) VT 1H NMR spectra of C1b, showing its transformation into C2b. Asterisks: solvent residual peak and Et2O. (B) 1H NMR spectrum of C2b. | ||
The causes for the inertness of certain ZnR2 (R = Me, CH2SiMe3 or CH2CMe2Ph) towards BIP ligands are not evident. Gibson suggested that the lack of reactivity of ZnMe2 could be related to the strength of the Zn–C bond in ZnMe2 as compared to ZnEt2 and higher alkyls.12 Our results confirm this relationship, as the Zn–C bond in unreactive Zn(CH2SiMe3)2 is particularly robust,15 while this is probably rather weak in dibenzyl or diallylzinc, which reacts readily with iPrBIP. The influence of the Zn–C bond strength on the reactivity of ZnR2 compounds towards BIP ligands seems to imply that the reaction is driven by the irreversible alkyl shift from the metal to the pyridine ring. However, we have shown that it is controlled by the formation of the ZnR2–BIP adduct C1, rather than by the cleavage of the Zn–C bond. Therefore, it can be concluded that the strength of the Zn–C bond has a critical influence on the ability of ZnR2 to bind BIP ligands. Early reports16 showed that the capability of ZnR2 compounds to bind bipyridyl (π-acceptor, like BIP) depends on the nature of R, increasing in the order Me<Et<iPr. This effect was attributed to the intensification of backdonation to bipyridyl as the R groups increase their electron-releasing capacity. More recently, it was suggested that backdonation involves electron transfer from the σ Zn–C bonds to the empty orbitals of the π-acceptor ligand.17 The complex ZnMe2(α-diimine) evidences the influence of this electronic flow in the lengthening of the Zn–C bond, compared to free ZnMe2.18 Thus, the observed relationship between the ability of Zn alkyls to bind BIP ligands and the Zn–C bond strength could stem from the fact that the more stable the σ bonding orbitals are, the lower is their capacity to donate electron density. This results in weaker backdonating capacity, hence in less stable metal–ligand interactions. Apart from controlling the complexation step, it should be stressed that the weakening of the σ M–C bond by charge transfer also favours cleavage and alkyl transfer to the heterocyclic ring.19
In summary, we have shown that the inherent ability of ZnR2 compounds to react with BIP ligands (specifically, iPrBIP) is strongly dependent on the nature of the R group and their ease in promoting the formation of intermediates [ZnR2(BIP)]. When R is benzyl or allyl, the reaction does take place and involves selective alkyl migration to the 4 position in the central pyridine ring, i.e., the same selectivity observed with dialkylmanganese(II) species. The resulting dihydropyridinate zinc derivatives are thermally stable, and in contrast with their Mn(II) analogues, they are not prone to aromatization by spontaneous hydrogen loss.
This work has been supported by the Spanish Government, Junta de Andalucía and the European Union (Projects CTQ2012-30962, FQM5074 and FEDER funds).
Notes and references
- (a) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS; (b) C. Bianchini, G. Giambastiani, I. G. Ríos, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2007, 250, 1391 CrossRef.
- A. M. Archer, M. W. Bouwkamp, M.-P. Cortez, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 4269 CrossRef CAS.
- (a) A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. Boyer, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567 CrossRef CAS; (b) A. M. Tondreau, C. C. H. Atienza, J. M. Darmon, C. Milsmann, H. M. Hort, K. J. Weller, S. A. Nye, K. M. Lewis, J. Boyer and J. G. P. Delis, Organometallics, 2012, 31, 4886 CrossRef CAS.
- (a) R. B. Bedford, M. Betham, D. W. Bruce, S. A. Davis, R. M. Frost and M. Hird, Chem. Commun., 2006, 1398 RSC; (b) O. Dayan, F. Dogan, K. Ismet and B. Cetinkaya, Synth. React. Inorg. Met.-Org. Chem., 2012, 40, 337 Search PubMed.
- M. D. Greenhalgh and S. P. Thomas, J. Am. Chem. Soc., 2012, 134, 11900 CrossRef CAS.
- (a) P. H. M. Budzelaar, B. de Bruin, A. W. Gal, K. Wieghardt and J. H. van Lenthe, Inorg. Chem., 2001, 40, 4649 CrossRef CAS; (b) Q. Knijnenburg, D. Hatterscheid, T. M. Kooistra and P. H. M. Budzelaar, Eur. J. Inorg. Chem., 2004, 1204 CrossRef CAS; (c) S. C. Bart, K. Chopek, E. Bill, M. W. Bowkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901 CrossRef CAS; (d) D. Zhu and P. H. M. Budzelaar, Organometallics, 2008, 27, 2699 CrossRef CAS.
- S. Blanchard, E. Derat, M. Desage-ElMurr, L. Fensterbank, M. Malacria and V. Mouriés-Mansuy, Eur. J. Inorg. Chem., 2012, 376 CrossRef CAS.
- C. M. Pérez, A. Rodríguez-Delgado, P. Palma, E. Álvarez, E. Gutiérrez-Puebla and J. Cámpora, Chem.–Eur. J., 2010, 16, 13834 CrossRef.
- J. Cámpora, C. M. Pérez, A. Rodríguez-Delgado, A. M. Naz, P. Palma and E. Álvarez, Organometallics, 2007, 26, 1104 CrossRef.
- (a) J. Cámpora, A. M. Naz, P. Palma, A. Rodríguez-Delgado, E. Álvarez, I. Tritto and L. Boggioni, Eur. J. Inorg. Chem., 2008, 1871 CrossRef; (b) A. Rodríguez-Delgado, J. Cámpora, A. M. Naz, P. Palma and M. L. Reyes, Chem. Commun., 2008, 5230 RSC; (c) J. Darmon, Z. R. Turner, E. Lobkovsky and P. J. Chirik, Organometallics, 2012, 31, 2275 CrossRef CAS.
- R. A. Layfield, Chem. Soc. Rev., 2008, 37, 1096 RSC.
- (a) D. Reardon, F. Conan, S. Gambarotta, J. P. A. Yap and Q. Wang, J. Am. Chem. Soc., 1999, 121, 9318 CrossRef CAS; (b) I. S. Korobkov, S. Gambarotta, J. P. A. Yap and P. H. M. Budzelaar, Organometallics, 2002, 21, 3088 CrossRef; (c) I. J. Blackmore, V. C. Gibson, P. B. C. Hitchcock, C. W. Rees, D. J. Williams and A. J. P. White, J. Am. Chem. Soc., 2005, 127, 6012 CrossRef CAS; (d) J. Scott, S. Gambarotta, I. Korobkov and P. H. M. Budzelaar, J. Am. Chem. Soc., 2005, 127, 13019 CrossRef CAS; (e) Q. Knijnenburg, J. M. M. Smits and P. H. M. Budzelaar, Organometallics, 2006, 25, 1036 CrossRef CAS; (f) I. Fernández, R. J. Trovitch, E. Lobkobsky and P. J. Chirik, Organometallics, 2008, 27, 109 CrossRef; (g) I. J. Blackmore, V. C. Gibson, P. B. C. Hitchcock, D. J. Williams and A. J. P. White, J. Am. Chem. Soc., 2008, 127, 6012 CrossRef.
- H. Lehmkuhl, I. Döring, R. McLane and H. Nehl, J. Organomet. Chem., 1981, 221, 1 CrossRef CAS.
- (a) M. W. Bowkamp, S. C. Bart, E. J. Hawrelak, R. J. Trovitch, E. Lobkovsky and P. J. Chirik, Chem. Commun., 2005, 3406 RSC; (b) J. Cámpora, A. M. Naz, P. Palma and E. Álvarez, Organometallics, 2005, 24, 4878 CrossRef.
- A. Haalan, C. Green, S. McGrady, A. J. Downs, E. Gallo, M. J. Lysall, J. Timberlake, A. V. Tutukin, H. V. Volden and K.-A. Østby, Dalton Trans., 2003, 4356 RSC.
- J. G. Noltes and J. Boersma, J. Organomet. Chem., 1967, 7, P6 CrossRef.
- (a) W. Kaim, Top. Curr. Chem., 1994, 169, 231 CrossRef CAS; (b) S. Hasenzahl, W. Kaim and T. Stahl, Inorg. Chim. Acta, 1994, 125, 23 CrossRef.
- M. Kaupp, H. Stoll, H. Preuss, W. Kaim, T. Stahl, G. van Koten, W. J. J. Smeets and A. L. Spek, J. Am. Chem. Soc., 1991, 113, 5606 CrossRef CAS.
- P. H. M. Budzelaar, Eur. J. Inorg. Chem., 2012, 530 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and crystallographic details. CCDC 934156. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc42798f |
| This journal is © The Royal Society of Chemistry 2013 |