 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate specificity of an oxygen dependent sulfoxide synthase in ovothiol biosynthesis†
Gabriel T. M.
Mashabela
ab and
Florian P.
Seebeck
*a
aDepartment for Chemistry, University of Basel, St. Johanns-Ring 19, Basel, Switzerland. E-mail: florian.seebeck@unibas.ch
bDepartment of Chemistry, University of Cape Town, Rondebosch, South Africa
First published on 4th July 2013
Abstract
OvoA is an iron(II) dependent sulfoxide synthase which catalyzes the first step in ovothiol A biosynthesis. This enzyme sulphurizes the C5 position of the imidazole side chain of L-histidine. We report the substrate specificity profile of this catalyst and present data which indicate that OvoA catalysis follows an thiol-ene type mechanism.
Ovothiol A (1, Fig. 1) is a thiohistidine derivative which has been discovered in sea urchin eggs1–3 and human pathogens such as Leishmania major and Trypanosoma cruzi.4–7 Because of its thiol function ovothiol A is characterized by a remarkably low pKa of 1.4,8 and an increased redox potential (−0.09 V vs. SHE) compared to glutathione (−0.26 V)8 or trypanothione (−0.24 V).9 These distinct parameters suggest that ovothiol A occupies functional niches in cellular redox homeostasis but its precise physiological roles are unknown.4–7,10–12
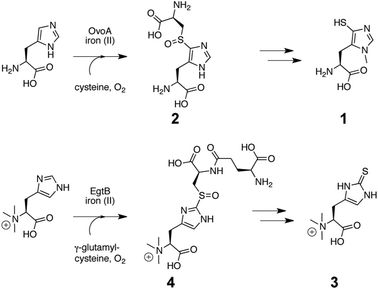 | ||
| Fig. 1 Biosynthesis of Ovothiol (1) in Erwinia tasmaniensis via 5-L-histidyl-L-cysteine sulfoxide intermediate (2).13 Bottom: biosynthesis of ergothioneine (1) in Mycobacterium smegmatis via a 2-N,N,N-α-trimethyl-L-histididyl-γ-L-glutamyl-L-cysteine sulfoxide intermediate (4).14 OvoA and EgtB are the first known sulfoxide synthases. | ||
Ovothiol A is biosynthesized from L-cysteine, L-histidine, molecular oxygen (O2) and S-adenosyl methionine (SAM) (Fig. 1).4–6 The key step in this pathway is oxidative insertion of a sulphur atom into the C5–H bond on the histidine side chain. Subsequent elimination of the L-cysteine derived carbon scaffold from intermediate 2 (Fig. 1) and reduction of the oxidized 5-thiohistidine complete the transfer of a sulphur atom from L-cysteine to L-histidine. Recently we characterized an iron(II) dependent enzyme, OvoA, from Erwinia tasmaniensis which mediates this unusual oxidative sulphur transfer.13 We also described an OvoA homolog from Mycobacterium smegmatis, EgtB, which is involved in ergothioneine biosynthesis (3, Fig. 1).14 The mycobacterial enzyme inserts a sulphur atom into the C2–H bond on the imidazole ring of α-N,N,N-trimethyl-L-histidine (4, Fig. 1).
The catalytic mechanism and the substrate scope of this novel class of non-heme iron enzymes are poorly understood. In the present report we demonstrate that OvoA catalyzes efficient in vitro sulphurization of L-histidine, D-histidine, 2-fluoro-L-histidine and compounds other than amino acids. In addition, we discuss indications that OvoA may catalyze C–S bond formation by a thiol-ene reaction mechanism,15 in which an OvoA generated L-cysteine thiyl radical attacks the unsaturated imidazole ring of L-histidine.
To initiate this study, we produced OvoA from E. tasmaniensis as previously reported.13 Typical OvoA reactions contained L-cysteine, L-histidine, 1 μM FeSO4, 1 mM ascorbate, 50 mM Tris HCl and 50 mM NaCl. The reactions were performed at 26 °C and were monitored by HPLC at 220 nm. Addition of either ascorbate or D-isoascorbate to the reaction mixture constituted a major improvement over our previous protocols13 because the antioxidants increased OvoA activity by nearly 100-fold (Fig. S1, ESI†). From a 800 mL reaction mixture containing 10 mg of OvoA, 1 mM L-histidine and 1 mM L-cysteine we were able to purify 100 mg of 5-L-histidyl-L-cysteine sulfoxide (2, Fig. 1, Fig. S2, ESI†), demonstrating than OvoA is able to deliver thiolated L-histidine at preparative scales.
OvoA catalysis is characterized by a kcat of 3.3 s−1 and a kcat/KM of 9.4 × 103 M−1 s−1 and 1.0 × 104 M−1 s−1 for L-cysteine and L-histidine respectively (Table 1, Fig. S3, ESI†). The pH dependence of kcat/KM,his follows a bell shaped curve with an activity maximum at pH 7.3 flanked by the kinetic pKas 6.8 and 8.0 (Fig. S4, ESI†). Alkaline pH limits activity due to a declining kcat. At lower pH KM,his becomes limiting. The lower kinetic pKa coincides with the pKa of L-histidine, suggesting that OvoA binds this substrate in deprotonated form.
| Sulfoxide product | kcat [s−1] | K M [M−1] | k cat/KM | |
|---|---|---|---|---|
| a Reactions containing 20 mM Tris HCl pH 8.0, 20 mM NaCl, 1 uM FeSO4, 0.28 uM OvoA, 2 mM tris(2-carboxyethyl)phosphine (TCEP) and 1 mM ascorbate were incubated at 26 °C. Product formation was monitored by HPLC at 220 nm. Products were identified by ESI MS (Table S1, ESI). Stated values are within an error margin of ±10% (Fig. S3 and S6, ESI). | ||||
| Sulphur donor | ||||
| L-Cysteine | 2 | 3.3 × 100 | 3.5 × 10−4 | 9.4 × 103 |
| Sulphur acceptor | ||||
| L-Histidine (5) | 2 | 3.4 × 100 | 3.4 × 10−4 | 1.0 × 104 |
| Histamine (7) | 26 | 3.6 × 10−1 | 2.3 × 10−4 | 1.6 × 103 |
| L-Histidinamide (6) | 25 | 5.7 × 10−1 | 3.1 × 10−4 | 1.8 × 103 |
| D-Histidine (9) | 28 and 29 | 7.8 × 10−1 | 6.7 × 10−4 | 1.2 × 103 |
| 4-Methyl imidazole (8) | 27 | n.a. | n.a. | 1.8 × 101 |
Using the same kinetic assay we then profiled the substrate specificity of OvoA. We previously determined that OvoA is highly specific for L-cysteine as a sulphur donor and does not accept other thiols such as D-cysteine, N-acetyl-L-cysteine, γ-L-glutamyl-L-cysteine, glutathione, or thiophosphate.13 In comparison, the specificity for the sulphur acceptor proved to be significantly broader. With the first compound series (6–14, Fig. 2) we probed the importance of the amino acid moiety in the sulphur acceptor, and using the second series (15–22, Fig. 2) examined whether OvoA accepts L-amino acids with alternative side chains.
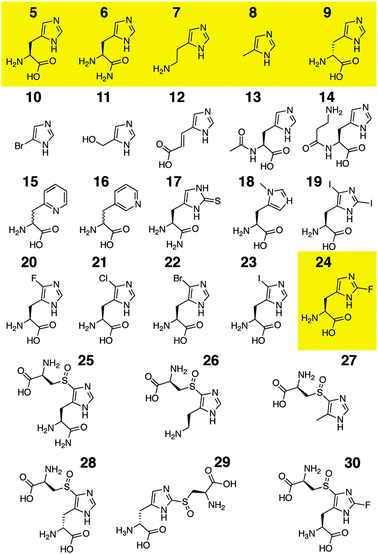 | ||
| Fig. 2 L-Histidine (5) and histidine analogs (6–24) tested for OvoA catalyzed turnover. Yellow: OvoA substrates. Sulfoxides 25–30 are ESI-MS identified OvoA products (Table S1, ESI†). | ||
L-Histidinamide (6) and histamine (7) are converted to products 25 and 26 only ten fold less efficiently than L-histidine. 4-Methyl imidazole is also a substrate demonstrating that the amino acid moiety of the sulphur acceptor is not essential for catalysis. On the other hand, we found no measurable turnover (kcat/KM < 1 M−1 s−1) of 4-bromo imidazole (10), 4(5)-(hydroxymethyl)imidazole (11), urocanic acid (12), N-α-acetyl-L-histidine (13) and carnosine (14).
Amino acid modifying enzymes are usually characterized by high enantioselectivity. Therefore we were surprised that D-histidine is fairly well tolerated as a substrate by OvoA (Table 1). However, detailed HPLC and 1H NMR analyses (Fig. S7, ESI†) identified the reaction product as a mixture of 63% 5-D-histidyl-L-cysteine sulfoxide (28) and 37% 2-D-histidyl-L-cysteine sulfoxide (29). We believe that the two regio-isomers result from two different binding modes of D-histidine to the OvoA active site in which either the C2 or the C5 position is presented to the reactive center. A preference for either binding mode may also explain the different regiospecificity for imidazole sulphurization in EgtB and OvoA (Fig. 1).
When we assayed OvoA with L-amino acids with imidazole derivatives as side chains we found much less substrate promiscuity. OvoA does not accept compounds 15–23 as substrates (Table 1). The remarkable exception in this series is 2-fluoro-L-histidine (24). Although an excellent L-histidine isostere,16 2-fluoro-L-histidine presents a much more acidic (pKa ≈ 1) and electron poor side chain.16 The redox potential of 2-fluoro-L-histidine is not known, but studies on indoles and phenols suggest that fluorination can increase the redox potential of aromatic moieties by 60–200 mV.17,18 Despite these changes OvoA converts 2-fluoro-L-histidine to 2-fluoro-5-L-histidyl-L-cysteine sulfoxide (30, HRMS: calcd 309.0669, found 309.0663) with almost the same efficiency as it converts L-histidine to 2 (Fig. S8, ESI†).
The apparent insensitivity of OvoA to the redox potential of the sulphur acceptor calls our earlier attempts at explaining the underlying catalytic mechanism into question.13 These proposals (1–3, Fig. 3) predicted sulfoxidation of the substrate L-cysteine as a requisite step to generate the enzyme bound oxo iron(IV) species (a) which then mediates C–S bond formation (b, c or d).13 This second step could proceed via homolytic cleavage of the imidazole C5–H bond (b, mechanism 1). Because the resulting sp2 radical is very unstable we would expect this to be the rate limiting step.19 Yet, when OvoA was assayed with deuterated D/L-histidine, we could not detect any substrate kinetic isotope effect (KIEsubstrate).13
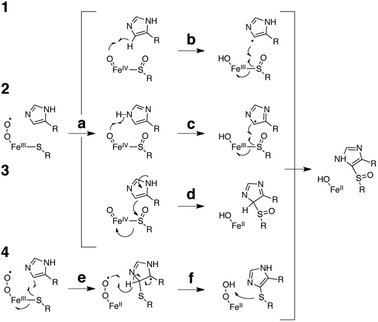 | ||
| Fig. 3 Proposed catalytic mechanism for OvoA catalyzed oxidative sulphur insertion into the imidazole C5–H bond of L-histidine. The current data are most consistent with mechanism 4. | ||
A second mechanism proposed one-electron oxidation of the imidazole ring coupled with deprotonation of the resulting imidazyl radical cation (c, mechanism 2). This reaction is still a difficult and likely rate limiting task for the oxo iron(IV) species.19 The absence of a significant solvent kinetic isotope effect (KIEsolvent = 1.2 ± 0.1, Fig. S5, ESI†) and the observation that the electron poor 2-fluoro-L-histidine is an efficient OvoA substrate suggest that this step is either not rate limiting or does not occur. The third mechanism which implicates the imidazole ring as a nucleophile (d, mechanism 3) is similarly inconsistent with efficient turnover of 2-fluoro-L-histidine, and with the absence of a KIEsolvent.
A fourth mechanism could explain the present observations more consistently: in this scheme the OvoA based iron(III)-superoxide complex generates a L-cysteine thiyl radical which attacks the imidazole ring (e), followed by rearomatisation (f). Subsequent sulfoxidation of the thioether restores the ferrous state of the enzyme and concludes the catalytic cycle. Because the imidazole ring serves as an electrophilic target for the nucleophilic thiyl radical it would not be surprising that 2-fluoro-L-histidine is a well tolerated substrate. According to this mechanism C–S bond formation depends only on the presence of an unsaturated carbon on the sulphur acceptor. This scheme is reminiscent of the thiol-ene reaction which relies on the ability of photo-generated thiyl radicals to attack olefins as a first step to thioether bond formation (Fig. 4).15 Given the broad scope of this reaction it seems possible that engineered or evolved sulfoxide synthases can be found which can sulphurize a broad range of unsaturated hydrocarbons.
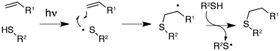 | ||
| Fig. 4 Proposed mechanism of the thiol-ene reaction.15 | ||
The reported data present OvoA from E. tasmaniensis as an efficient catalyst allowing in vitro preparation of the sulphurized product (2, Fig. 1) on a 100 mg scale. The substrate specificity profile suggests that OvoA does not require an amino acid moiety on the sulphur acceptor. OvoA converts D-histidine into a mixture of C2 and C5 modified products indicating that product specificity is purely a function of substrate positioning in the active site. Finally, our observation that OvoA accepts 2-fluoro-L-histidine as an efficient substrate, coupled with the absence of KIEsolvent or KIEsubstrate, point towards a catalytic mechanism which views the sulphur acceptor as the passive target of an iron(III)-superoxide generated thiyl radical. In combination, these properties present OvoA as a promising scaffold for the engineering of tailor made sulphur transferases.
The authors thank Kenneth Kirk for the generous gift of a precious sample of 2-fluoro-L-histidine and Ali Alkaabi for recording HRMS data. G.T.M.M. is a recipient of a Swiss government fellowship for excellence; F.P.S. is supported by the “Professur für Molecular Bionics”, and by the Swiss National Science Foundation.
Notes and references
- A. Palumbo, M. Dischia, G. Misuraca and G. Prota, Tetrahedron Lett., 1982, 23, 3207–3208 CrossRef CAS.
- E. Turner, R. Klevit, P. B. Hopkins and B. M. Shapiro, J. Biol. Chem., 1986, 261, 3056–3063 Search PubMed.
- E. Turner, L. J. Hager and B. M. Shapiro, Science, 1988, 242, 939–941 CrossRef CAS.
- D. J. Steenkamp and H. S. C. Spies, Eur. J. Biochem., 1994, 223, 43–50 CrossRef CAS.
- D. J. Steenkamp, D. Weldrick and H. S. C. Spies, Eur. J. Biochem., 1996, 242, 557–566 CAS.
- R. N. Vogt, H. S. C. Spies and D. J. Steenkamp, Eur. J. Biochem., 2001, 268, 5229–5241 CrossRef CAS.
- M. R. Ariyanayagam and A. H. Fairlamb, Mol. Biochem. Parasitol., 2001, 115, 189–198 CrossRef CAS.
- K. H. Weaver and D. L. Rabenstein, J. Org. Chem., 1995, 60, 1904–1907 CrossRef CAS.
- A. H. Fairlamb and A. Cerami, Annu. Rev. Microbiol., 1992, 46, 695–729 CrossRef CAS.
- R. L. Krauth-Siegel, H. Bauer and H. Schirmer, Angew. Chem., Int. Ed., 2005, 44, 690–715 CrossRef CAS.
- C. Jacob, Nat. Prod. Rep., 2006, 23, 851–863 RSC.
- R. L. Krauth-Siegel and A. E. Leroux, Antioxid. Redox Signaling, 2012, 17, 583–607 CrossRef CAS.
- A. Braunshausen and F. P. Seebeck, J. Am. Chem. Soc., 2011, 133, 1757–1759 CrossRef CAS.
- F. P. Seebeck, J. Am. Chem. Soc., 2010, 132, 6632–6633 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- D. S. Wimalasena, J. C. Cramer, B. E. Janowiak, S. J. Juris, R. A. Meinyk, D. E. Anderson, K. L. Kirk, R. J. Collier and J. G. Bann, Biochemistry, 2007, 46, 14928–14936 CrossRef CAS.
- T. Liu, P. R. Callis, B. H. Hesp, M. de Groot, W. J. Buma and J. Broos, J. Am. Chem. Soc., 2005, 127, 4104–4113 CrossRef CAS.
- M. R. Seyedsayamdost, S. Y. Reece, D. G. Nocera and J. Stubbe, J. Am. Chem. Soc., 2006, 128, 1569–1579 CrossRef CAS.
- E. A. Bushnell, G. B. Fortowsky and J. W. Gauld, Inorg. Chem., 2013, 51, 13351–13356 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S8, Table S1, and detailed experimental procedures. See DOI: 10.1039/c3cc42594k |
| This journal is © The Royal Society of Chemistry 2013 |