 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
J- vs. H-type assembly: pentamethine cyanine (Cy5) as a near-IR chiroptical reporter†
Larysa I.
Markova
ab,
Vladimir L.
Malinovskii
a,
Leonid D.
Patsenker
b and
Robert
Häner
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: robert.haener@ioc.unibe.ch; Tel: +41 31 631 4382
bDepartment of Organic Luminophores and Dyes, “Institute for Single Crystals”, Nat. Acad. Sci. Ukraine, 60, Lenin Ave., Kharkiv 61001, Ukraine. E-mail: patsenker@isc.kharkov.com; Tel: +380-57-341-02-72
First published on 24th April 2013
Abstract
The DNA-enabled dimerization of pentamethine cyanine (Cy5) dyes was studied by optical methods. The value of cyanine as a chiroptical reporter using a monomer-to-dimer switch is demonstrated. The specific shape of the CD signal and its high intensity are a result of J-type assembly.
The use of chiroptical labels is usually not considered as a first line method for biological investigations because of their relatively low sensitivity compared to other optical techniques.1 On the other hand, circular dichroism (CD) strongly depends on the three-dimensional arrangement of the interacting units and may, therefore, provide information not accessible by other methods. Chiroptical reporters are probes of structural information2–4 with special value in applications of exciton-coupled CD for the characterization of complex materials, including the determination of absolute configuration.2,5 Due to their excellent absorbance and fluorescence properties, cyanine dyes are a class of compounds that find use as sensitive labels for biology related applications, such as the study of DNA and proteins, including single molecule spectroscopy in live cells.6–8 Herein, we demonstrate the value of cyanine dyes as chiroptical reporters in the visible to near-IR spectral range. Oligonucleotide–cyanine conjugates reveal a high susceptibility to monomer-to-dimer transitions that are observed by asymmetric exciton couplets due to J-aggregate formation.
We followed our general design of oligonucleotide conjugates with internally positioned artificial building blocks using the phosphoramidite approach (Scheme 1).9–11 Due to the widespread interest in cyanine derivatives as labels and probes, the pentamethine cyanine (Cy5) phosphoramidite (abbreviated herein as Cy) is commercially available. Oligonucleotides ON1, ON2 and unmodified references ON3, ON4 were obtained from custom suppliers (ESI†). The recently reported squaraine (Sq) modified oligonucleotide ON512 served as a reference oligomer and also as a counter strand in mixed hybrid experiments.
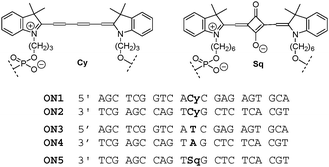 | ||
| Scheme 1 Cyanine- and squaraine-modified oligomers and reference oligonucleotides (ON1–ON5). | ||
An overview of the absorbance and fluorescence data of cyanine modified oligonucleotides is presented in Table 1 and Fig. 1. Absorbance in the area up to 300 nm is mainly due to the nucleobases, whereas Cy and Sq absorption is manifested in the visible to near-IR area (500–700 nm). A key observation is the change in the shapes of the Cy and Sq spectra in the single strands (corresponding to the spectra of the monomer dyes) and those of the Cy–Cy and Cy–Sq assemblies in the hybrids. The development of a new, red-shifted absorption band of the ON1*ON2 hybrid compared to the monomer (668 vs. 648 nm) is characteristic of J-aggregation of the Cy dyes,13–15 a J-dimer in this case. In contrast, in the mixed assembly Cy–Sq (ON1*ON5) the major band is blue-shifted (591 nm) compared to the band of each monomer, revealing preferential H-type coupling.
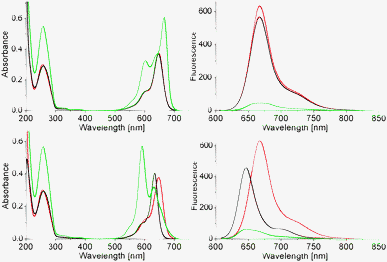 | ||
| Fig. 1 Top: absorption (left) and fluorescence (right) spectra of single strands ON1 (red) and ON2 (black), and their hybrid ON1*ON2 (green); bottom: absorption and fluorescence spectra of ON1 (red), ON5 (black) and hybrid ON1*ON5 (green, 1.5 μM single strand conc., 10 mM phosphate, pH = 7.4, 100 mM NaCl). | ||
Fluorescence data show that either form of assembly results in a substantial decrease in the Cy signal (>90%, Table 1 and Fig. 1). The excitation spectrum (ESI,† Fig. S4) has a maximum at 650 nm revealing that the remaining fluorescence signal of the ON1*ON2 hybrid originates from a monomer type chromophore. Variable temperature absorption spectra of single strand ON2 (ESI,† Fig. S5) disclose no specific interactions of the cyanine with other parts of the same oligomer (negligible deviations in absorbance). A decrease in fluorescence is observed with rising temperature (70% reduction, 20 → 80 °C), which is in agreement with the thermochromic behaviour described for cyanines.16,17
The higher tendency of cyanines to form J-type assemblies15,17 compared to other organic molecules is well documented. H- and J-assembly of cyanines in crystals and extended aggregates on surfaces or in solution, including prolonged assemblies on DNA and other polymers as a template, has been reported.18–22 The precise composition of J-assemblies is often ill-defined and the number of units participating in coherent J-coupling is uncertain.15,23–25 The oligonucleotide-driven approach enables a molecularly defined constitution of the assembly. Spectroscopic changes distinctly show a transition between monomeric and dimeric forms of Cy molecules. The development of a J-band (Δλ = 20 nm) upon formation of a Cy-dimer is evident upon titration of ON1 to ON2 (Fig. 2). More accurately, assembly of Cy within the ON1*ON2 hybrid is accompanied by the development of two main bands consistent with Davydov exciton splitting21,22 of the monomer band (M, λmax = 648 nm) into two levels, at lower (λmax = 668 nm, J-band) and higher (λmax = 601 nm, H-band) energies, which reveals an “oblique” orientation of the chromophores.3,26 This arrangement of the cyanines is crucial for the appearance of strong CD effects (see below).
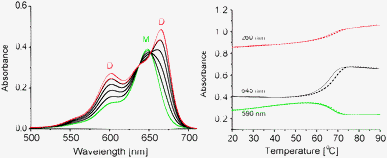 | ||
Fig. 2 Left: development of exciton splitting (J- and H-bands) upon Cy–Cy assembly; right: thermal denaturation of ON1*ON2; cooling (–), heating (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ); (Tm = 67 °C; M: Cy monomer, D: Cy dimer); single strand conc. 1.5 μM, conditions: see Fig. 1. ); (Tm = 67 °C; M: Cy monomer, D: Cy dimer); single strand conc. 1.5 μM, conditions: see Fig. 1. | ||
The melting temperature (Tm) of the ON1*ON2 hybrid (67 °C, Fig. 2) is 4 °C lower than that of the unmodified duplex ON3*ON4 (71 °C). This is comparable to the effect of a nucleobase mismatch or an abasic site in a DNA duplex27 and in contrast to the effects of other modifications of the same motif, such as pyrene,28 PDI29 or fluorene.30 The latter modifications increase duplex stability due to stacking in a presumed face-to-face H-type fashion.
The most inspiring results came from circular dichroism (CD) experiments. The CD spectrum of the ON1*ON2 hybrid is shown in Fig. 3 (left). A very strong intensity and a robust, specific signal shape are manifested. Several Cotton effects are evident: 668 nm (Δε + 250), 633 (Δε − 50) and 601 nm (Δε − 80). CD effects are absent or negligible in the corresponding single strands, which is attributed to the flexibility of the single strands. Signal shape and intensity are functions of dipolar strength of the electronic transitions and the mutual arrangement of the interacting transition dipoles, i.e. they depend on both, energetic and geometric factors. Usually, an exciton coupled CD of H-assembled chromophores is of symmetrical shape with positive and negative couplets of comparable intensities; it can be reasonably described by considering the main electronic transition only.31,32 The complexity of the CD couplet in the ON1*ON2 hybrid is due to the J-type dimer formation which leads to a substantial increase in the oscillator strength of the low energy band and its narrow character. The simulation of the observed spectrum has the best fit when the monomer band (648 nm) gives an asymmetric exciton couplet (+668 nm, −633 nm, with amplitude A = +300 and width W = 35 nm) and the other bands (601 and 556 nm) are (mono-directional) negative signals (see ESI,† S9). The CD spectrum is asymmetric in shape with a maximum that corresponds to the λmax of the J-band, and a crossing point at 645 nm, which is close to the λmax of the monomer absorbance. Notably, the overall integral intensity 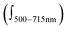 of the positive (+) signal is significantly enhanced compared to the negative (−) one,
of the positive (+) signal is significantly enhanced compared to the negative (−) one, 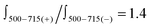 . Non-equivalence in the intensities of exciton couplets of the main transition
. Non-equivalence in the intensities of exciton couplets of the main transition 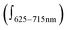 is very pronounced and manifested in a 6-fold more intense positive couplet (
is very pronounced and manifested in a 6-fold more intense positive couplet (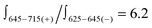 ; Fig. 3 and ESI,† S9). Quantitative characterization of signal asymmetry is possible using g-factor values,2 which gives g668/g633 = 4.2 and g668/g601 = 2.6 (see ESI†).
; Fig. 3 and ESI,† S9). Quantitative characterization of signal asymmetry is possible using g-factor values,2 which gives g668/g633 = 4.2 and g668/g601 = 2.6 (see ESI†).
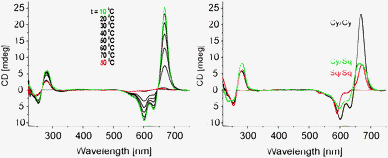 | ||
| Fig. 3 Left: CD spectra of the ON1*ON2 hybrid in the temperature range 10–80 °C; right: CD spectra of different assemblies (20 °C); single strand conc. 1.5 μM, conditions: see Fig. 1. | ||
The Sq dye (Scheme 1) has similar optical properties to Cy (λmaxCy = 645 nm, εCy = 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1; λmaxSq = 635 nm, εSq = 258000 M−1 cm−1) and was used as a reference compound and a counterpart in coupling experiments with non-identical chromophores. Sq–Sq excitonic interactions in the corresponding DNA hybrids12 show a significantly more populated transition at higher energy (H-aggregation) and a symmetrical CD signal (Fig. 3, right). Non-degenerate interaction of Cy–Sq within ON1*ON5 also results in a higher fraction of H-type coupling (λmax = 591 nm, see absorption changes, Fig. 1) and gives a CD of complex shape with an overall broadening of bands. Most significantly, the CD signal intensity of the low energy band of Cy–Sq is at the level of that of Sq–Sq (H-type coupling,12Fig. 3, right), but is significantly lower than that observed for Cy–Cy (J-type) coupling. In absolute values, the signal intensity (I) of the Cy–Cy pair at 668 nm was 3.5 times higher than that of 20 natural oligonucleotide base pairs (bp) at 280 nm leading to a ratio ICyCy/Ibp ≅ 70. Semi-quantitative comparison of chiroptical labels in a DNA framework can be performed. Since the CD spectrum shows an averaged signal in the DNA region, the value per base pair should be approximately the same for a B-DNA of any given length, composition and base sequence. A value of Δε280 = 3–4 M−1 cm−1 per base-pair is commonly observed for a B-type DNA under physiological conditions.33 Our data are consistent with this value: Δε280(B-DNA) ∼ Δε280(ON1*ON2) ∼ Δε280(ON3*ON4) ∼ 3.5 M−1 cm−1 per base pair is found, which gives Δε668(Cy–Cy)/Δε280(bp) ≅ 70. This ratio can be used as a rough but useful value for a first approximation in comparison of CD data of modified oligonucleotides that are increasingly appearing,34–44 and as a reference value for the design of DNA-based chiroptical reporters.4,5 The remarkably high sensitivity of the Cy chiroptical label is further demonstrated by the titration experiment shown in Fig. 4: a 7.4 × 10−8 M oligonucleotide concentration is easily detected in a standard 1 cm cell (see ESI†).
000 M−1 cm−1; λmaxSq = 635 nm, εSq = 258000 M−1 cm−1) and was used as a reference compound and a counterpart in coupling experiments with non-identical chromophores. Sq–Sq excitonic interactions in the corresponding DNA hybrids12 show a significantly more populated transition at higher energy (H-aggregation) and a symmetrical CD signal (Fig. 3, right). Non-degenerate interaction of Cy–Sq within ON1*ON5 also results in a higher fraction of H-type coupling (λmax = 591 nm, see absorption changes, Fig. 1) and gives a CD of complex shape with an overall broadening of bands. Most significantly, the CD signal intensity of the low energy band of Cy–Sq is at the level of that of Sq–Sq (H-type coupling,12Fig. 3, right), but is significantly lower than that observed for Cy–Cy (J-type) coupling. In absolute values, the signal intensity (I) of the Cy–Cy pair at 668 nm was 3.5 times higher than that of 20 natural oligonucleotide base pairs (bp) at 280 nm leading to a ratio ICyCy/Ibp ≅ 70. Semi-quantitative comparison of chiroptical labels in a DNA framework can be performed. Since the CD spectrum shows an averaged signal in the DNA region, the value per base pair should be approximately the same for a B-DNA of any given length, composition and base sequence. A value of Δε280 = 3–4 M−1 cm−1 per base-pair is commonly observed for a B-type DNA under physiological conditions.33 Our data are consistent with this value: Δε280(B-DNA) ∼ Δε280(ON1*ON2) ∼ Δε280(ON3*ON4) ∼ 3.5 M−1 cm−1 per base pair is found, which gives Δε668(Cy–Cy)/Δε280(bp) ≅ 70. This ratio can be used as a rough but useful value for a first approximation in comparison of CD data of modified oligonucleotides that are increasingly appearing,34–44 and as a reference value for the design of DNA-based chiroptical reporters.4,5 The remarkably high sensitivity of the Cy chiroptical label is further demonstrated by the titration experiment shown in Fig. 4: a 7.4 × 10−8 M oligonucleotide concentration is easily detected in a standard 1 cm cell (see ESI†).
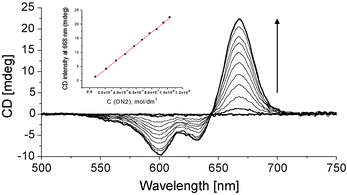 | ||
| Fig. 4 Duplex formation by stepwise addition (arrow) of ON2 to ON1 (starting conc. 1.5 μM); first addition of ON2: 7.4 × 10−8 M. Inset: concentration dependence of signal intensity at 668 nm. | ||
In conclusion, using a derivative of the widely used chromophore Cy5, we have demonstrated the value of cyanine dyes as chiroptical reporters. Cy–Cy interaction leads to intense CD effects in a DNA duplex. The origin of the high signal intensity resides in the formation of a J-type dimeric assembly that exhibits an asymmetric exciton coupled CD signal with a significant enhancement of one of the couplets. This type of aggregation is well known for cyanine dyes,16–20 however, to the best of our knowledge, was not applied so far in the design of chiroptical labels using a monomer-to-dimer switch.
Notes and references
- G. Snatzke, in Circular Dichroism – Principles and Applications, ed. N. Berova, K. Nakanishi and R. W. Woody, Wiley-VCH, New York, 2000, pp. 1–35 Search PubMed.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- S. E. Boiadjiev and D. A. Lightner, Monatsh. Chem., 2005, 136, 489–508 CrossRef CAS.
- G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603–4625 RSC.
- L. Patsenker, A. Tatarets, O. Kolosovaa, O. Obukhova, Y. Povrozin, I. Fedyunyayeva, I. Yermolenko and E. Terpetschnig, Ann. N. Y. Acad. Sci., 2008, 1130, 179–187 CrossRef CAS.
- B. Oswald, L. Patsenker, J. Duschl, H. Szmacinski, O. S. Wolfbeis and E. Terpetschnig, Bioconjugate Chem., 1999, 10, 925–931 CrossRef CAS.
- M. S. Goncalves, Chem. Rev., 2009, 109, 190–212 CrossRef CAS.
- S. M. Langenegger and R. Häner, Helv. Chim. Acta, 2002, 85, 3414–3421 CrossRef CAS.
- V. L. Malinovskii, D. Wenger and R. Häner, Chem. Soc. Rev., 2010, 39, 410–422 RSC.
- Y. N. Teo and E. T. Kool, Chem. Rev., 2012, 112, 4221–4245 CrossRef CAS.
- L. I. Markova, V. L. Malinovskii, L. D. Patsenker and R. Häner, Org. Biomol. Chem., 2012, 10, 8944–8947 CAS.
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS.
- G. Scheibe, Angew. Chem., 1936, 49, 563 CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Moeller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- A. Kulinich and V. A. Ishchenko, Russ. Chem. Rev., 2009, 78, 141–164 CrossRef CAS.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2011 CrossRef CAS.
- K. C. Hannah and B. A. Armitage, Acc. Chem. Res., 2004, 37, 845–853 CrossRef CAS.
- M. M. Wang, G. L. Silva and B. A. Armitage, J. Am. Chem. Soc., 2000, 122, 9977–9986 CrossRef CAS.
- S. Kirstein and S. Daehne, Int. J. Photoenergy, 2006, 2006, 20363 Search PubMed.
- C. Peyratout, E. Donath and L. Daehne, J. Photochem. Photobiol., A, 2001, 142, 51–57 CrossRef CAS.
- C. Peyratout and L. Daehne, Phys. Chem. Chem. Phys., 2002, 4, 3032–3039 RSC.
- I. G. Scheblykin, O. Y. Sliusarenko, L. S. Lepnev, A. G. Vitukhnovsky and M. Van der Auweraer, J. Phys. Chem. B, 2001, 105, 4636–4646 CrossRef CAS.
- G. Calzaferri, R. Meallet-Renault, D. Brühwiler, R. Pansu, I. Dolamic, T. Dienel, P. Adler, H. R. Li and A. Kunzmann, ChemPhysChem, 2011, 12, 580–594 CrossRef CAS.
- V. Sundstrom, T. Pullerits and R. van Grondelle, J. Phys. Chem. B, 1999, 103, 2327–2346 CrossRef.
- M. Kasha, Radiat. Res., 1963, 20, 55–70 CrossRef CAS.
- S. M. Langenegger and R. Häner, ChemBioChem, 2005, 6, 848–851 CrossRef CAS.
- H. Bittermann, D. Siegemund, V. L. Malinovskii and R. Häner, J. Am. Chem. Soc., 2008, 130, 15285–15287 CrossRef CAS.
- N. Bouquin, V. L. Malinovskii and R. Häner, Chem. Commun., 2008, 1974–1976 RSC.
- D. Wenger, V. L. Malinovskii and R. Häner, Chem. Commun., 2011, 47, 3168–3170 RSC.
- N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism – Principles and Applications, Wiley-VCH, New York, 2nd edn, 2000 Search PubMed.
- S. Superchi, E. Giorgio and C. Rosini, Chirality, 2004, 16, 422–451 CrossRef CAS.
- W. C. Johnson, in Circular Dichroism – Principles and Applications, ed. N. Berova, K. Nakanishi and R. W. Woody, Wiley-VCH, New York, 2000, pp. 703–718 Search PubMed.
- L. Bethge, D. V. Jarikote and O. Seitz, Bioorg. Med. Chem., 2008, 16, 114–125 CrossRef CAS.
- A. Mammana, G. Pescitelli, T. Asakawa, S. Jockusch, A. G. Petrovic, R. R. Monaco, R. Purrello, N. J. Turro, K. Nakanishi, G. A. Ellestad, M. Balaz and N. Berova, Chem.–Eur. J., 2009, 15, 11853–11866 CrossRef CAS.
- H. Kashida and H. Asanuma, Phys. Chem. Chem. Phys., 2012, 14, 7196–7204 RSC.
- R. Varghese and H. A. Wagenknecht, Chem. Commun., 2009, 2615–2624 RSC.
- M. Nakamura, Y. Murakami, K. Sasa, H. Hayashi and K. Yamana, J. Am. Chem. Soc., 2008, 130, 6904–6905 CrossRef CAS.
- I. Bouamaied, T. Nguyen, T. Ruhl and E. Stulz, Org. Biomol. Chem., 2008, 6, 3888–3891 CAS.
- M. Endo, M. Fujitsuka and T. Majima, J. Org. Chem., 2008, 73, 1106–1112 CrossRef CAS.
- S. M. Biner, D. Kummer, V. L. Malinovskii and R. Häner, Org. Biomol. Chem., 2011, 9, 2628–2633 CAS.
- T. S. Kumar, A. S. Madsen, M. E. Ostergaard, J. Wengel and P. J. Hrdlicka, J. Org. Chem., 2008, 73, 7060–7066 CrossRef CAS.
- F. D. Lewis, L. G. Zhang, X. Y. Liu, X. B. Zuo, D. M. Tiede, H. Long and G. C. Schatz, J. Am. Chem. Soc., 2005, 127, 14445–14453 CrossRef CAS.
- V. V. Filichev and E. B. Pedersen, in Wiley Encycl. Chem. Biol., ed. T. P. Begley, Wiley, Hoboken, 2009, vol. 1, pp. 493–524 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and analytical details; additional spectra. See DOI: 10.1039/c3cc42103a |
| This journal is © The Royal Society of Chemistry 2013 |