 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The long-lived electron transfer state of the 2-phenyl-4-(1-naphthyl)quinolinium ion incorporated into nanosized mesoporous silica–alumina acting as a robust photocatalyst in water†
Yusuke
Yamada
a,
Akifumi
Nomura
a,
Kei
Ohkubo
a,
Tomoyoshi
Suenobu
a and
Shunichi
Fukuzumi
*ab
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370
bDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
First published on 15th March 2013
Abstract
A simple electron donor–acceptor linked dyad, the 2-phenyl-4-(1-naphthyl)quinolinium ion (QuPh+–NA), was incorporated into nanosized mesoporous silica–alumina to form a composite, which is highly dispersed in water and acts as an efficient and robust photocatalyst for the reduction of O2 by oxalate to produce hydrogen peroxide with a quantum yield of 10%.
Artificial photosynthesis attracts many researchers to realize a sustainable society depending mainly on solar energy.1 A variety of electron donor–acceptor linked molecules, which mimic charge-separation processes in the photosynthetic reaction centre, have been utilized in photocatalytic systems to produce high-energy chemicals.2,3 The donor–acceptor linked molecules form an electron-transfer (ET) state upon photoirradiation in reaction media, usually in organic solvents.4–6 Even if an electron donor–acceptor linked molecule exhibits a long-lived ET state in the isolated state, e.g., in frozen media, the lifetime of its ET state in a solution is much shorter than that in the isolated state, because of facile intermolecular back electron transfer.6
To avoid such an intermolecular event in solution, electron donor–acceptor linked molecules have been isolated on a metal oxide support. For example, 9-mesityl-10-methylacridinium ion (Acr+-Mes), which forms an ET state upon photoirradiation, has been supported on mesoporous silica–alumina by ion exchange, exhibiting the ET state lifetime longer than a second when suspended in acetonitrile (MeCN) at room temperature.7 Among the reaction media used for chemical reactions, water is the most commonly used solvent owing to its abundance, ubiquitous nature and environmentally benign properties. Thus, efficient photocatalytic processes using a long-lived ET state in water are highly desired. However, organic donor–acceptor linked molecules are yet to be employed as photocatalysts in water because of their insolubility in water.
We report herein the incorporation of the 2-phenyl-4-(1-naphthyl)quinolinium ion (QuPh+–NA), which also forms the ET state acting as a strong oxidant and reductant (Ered = 1.87 V and Eox = −0.90 V vs. SCE in MeCN) upon photoirradiation,5 into nanosized mesoporous silica–alumina with a spherical shape (sAlMCM-41) by cation exchange and utilization of the composite (QuPh+–NA@sAlMCM-41) as a photocatalyst in aqueous media. The composite affords the extremely long-lived ET state (QuPh˙–NA˙+@sAlMCM-41) upon photoirradiation even in the presence of water. Thus, the photocatalytic activity of the composite was investigated for O2 reduction by the oxalate anion to produce H2O2 in water. Oxalate is converted into two equivalents of CO2 after the reaction, therefore, pure H2O2 in water can be easily obtained by just removing the catalytic nanocomposite by filtration or sedimentation.
Na+-exchanged sAlMCM-41 has been prepared according to a reported method with modifications.8 TEM images of the synthesized Na+-exchanged sAlMCM-41 are displayed in Fig. 1a and Fig. S1 in ESI.† N2 adsorption and desorption isotherms for Na+-exchanged sAlMCM-41 afforded a Brunauer–Emmett–Teller (BET) surface area of 520 m2 g−1 with a pore diameter of 1.8 nm determined by the Barrett–Joyner–Halenda (BJH) method (Fig. S2 in ESI†). The Na+-exchanged sAlMCM-41 was ion-exchanged with QuPh+–NA to prepare the QuPh+–NA@sAlMCM-41 composite. Because the molecular size of QuPh+–NA is small compared with the pore size of sAlMCM-41, cation exchange with QuPh+–NA occurs spontaneously upon mixing Na+-exchanged sAlMCM-41 with QuPh+–NA(ClO4−) in MeCN as observed by UV-vis absorption of the supernatant (Fig. S3, ESI†). The amount of adsorbed QuPh+–NA was determined by the decrease in absorption of the solution at 333 nm owing to QuPh+–NA. The number of Al atoms on the surface of sAlMCM-41 estimated from the used amount of Al source was 1.05 × 10−3 mol g−1, thus, 7.9% of all the Na+-sites was occupied by QuPh+–NA. The diffused reflectance UV-vis spectrum of the QuPh+–NA@sAlMCM-41 composite shown in Fig. 1b (red solid line) shows that the positions of peaks and shoulders of the spectrum are quite similar to those of the QuPh+–NA ion in MeCN (black dotted line in Fig. 1b).
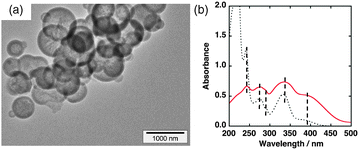 | ||
| Fig. 1 (a) Transmission electron microscope (TEM) image of Na+-exchanged sAlMCM-41. (b) Diffuse reflectance UV-vis spectrum of the QuPh+–NA@sAlMCM-41 composite (red solid line) compared with the UV-vis absorption spectrum of the QuPh+–NA ion in MeCN (black dotted line). Positions of peaks and shoulders are indicated by broken lines. | ||
Photoirradiation of the QuPh+–NA@sAlMCM-41 composite using a 1000 W high-pressure mercury lamp through a UV-light cutting filter (λ > 340 nm) results in the formation of the ET state (QuPh˙–NA˙+@sAlMCM-41) via photoinduced electron transfer from the naphthalene (NA) moiety to the singlet excited state of the quinolinium ion (QuPh+) moiety as evidenced by EPR measurements. An EPR signal at g = 2.0031 appearing upon photoirradiation (Fig. 2a) assures formation of a radical species. The decrease in the EPR signal intensities observed by cutting off the light obeyed first-order kinetics for a prolonged time after 20 s as shown in Fig. 2b (red dotted line), revealing the intramolecular back electron transfer of the ET state of QuPh+–NA. Based on the rate constant of the decay curve, the lifetime of the ET state is longer than 190 s at 316 K. The ET state lifetime in the presence of water significantly decreased as compared with that observed in the absence of water (Fig. 2b, black solid line). Water vapour was adsorbed on the composite at 313 K under reduced pressure for several hours, because the presence of bulk water strongly interferes the observation of EPR signals. The EPR signal intensity oscillated by the intermittent photoirradiation for 2 seconds followed by 48 s in the dark as indicated in Fig. 2c. The similar signal intensity obtained at each cycle resulted from the photorobustness of the composite. An Eyring plot in Fig. 2d for the rate constant of intramolecular back electron transfer in the ET state of the composite indicates that the activation enthalpy (ΔH‡) of the composite was determined to be 12(±2) kcal mol−1. A large ΔH‡ value [20 (±1) kcal mol−1] has also been reported for Acr+-Mes supported on AlMCM-41 in MeCN,7 resulting from a large driving force for the back electron transfer in the Marcus inverted region,9 because of decreased solvation in the pores of sAlMCM-41.
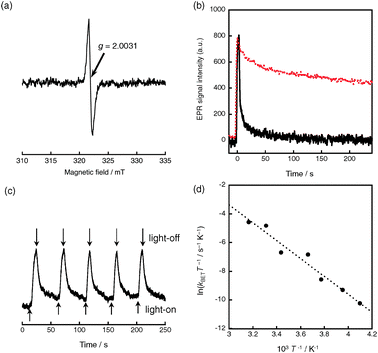 | ||
| Fig. 2 (a) EPR spectrum of the QuPh+–NA@sAlMCM-41 composite in the presence of adsorbed water under photoirradiation using a high-pressure mercury lamp and a UV cut-off filter (λ > 340 nm). (b) Time profile of the EPR signal intensity for QuPh˙–NA˙+@sAlMCM-41 in the absence (red dotted) or presence (black solid) of water at 316 K. (c) Time profile of the EPR signal intensity for QuPh˙–NA˙+@sAlMCM-41 in the presence of water upon intermittent photoirradiation for 2 seconds followed by 48 seconds in the dark at 316 K. (d) Plot of ln(kBETT−1) vs. T−1 for intramolecular back electron transfer of QuPh+–NA@sAlMCM-41 in the presence of water. The water-adsorbed sample was prepared by exposure of QuPh+–NA@sAlMCM-41 to water vapour at 313 K overnight under reduced pressure. The time profile at each temperature is indicated in Fig. S4 in ESI.† | ||
The QuPh+–NA@sAlMCM-41 composite was examined as a photocatalyst for the production of H2O2 in an aqueous solution. H2O2 can be simply prepared by reduction of atmospheric O2 by oxalate, which is capable of acting as a strong reductant (Eo = −0.48 V vs. NHE).10 Oxalate can be easily found in the soil and leaves of various vegetables at a high content of ∼1 wt%.11 Nature utilizes oxalate as an electron donor for producing H2O2 in the enzymatic active centre of oxalate oxidase, which catalyses the oxidation of oxalate, reduction of O2 to H2O2 and formation of two moles of CO2 [eqn (1)].12 In this process, CO2 is solely produced as a
| (COO−)2 + 2H+ + O2 → 2CO2 + H2O2 | (1) |
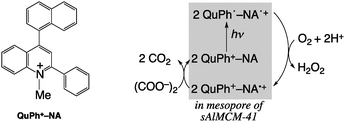 | ||
| Scheme 1 Chemical structure of QuPh+–NA and overall catalytic cycle for H2O2 production. | ||
The photocatalytic H2O2 production has been performed by photoirradiation (λ > 340 nm) of an oxygen-saturated aqueous suspension (1.3 mM, 2.0 mL) containing QuPh+–NA@sAlMCM-41 (QuPh+–NA, 0.22 mM) and oxalate (100–400 mM). Fig. 3a shows the time courses of H2O2 production in the photocatalytic reaction. The initial H2O2 formation rate (1 h) increased from 1.6 μmol h−1 to 2.5 and 3.5 μmol h−1 upon increasing the concentration of oxalate from 100 mM to 200 and 400 mM in the reaction solution. The pH of the reaction solution was controlled by changing the ratio of the amount of oxalic acid to that of disodium oxalate keeping the concentration of oxalate constant (200 mM) as shown in Fig. 3b. The initial H2O2 formation rate (1 h) reached the maximum around pH 4.5. A further increase or decrease in pH decelerated the H2O2 formation. At lower pH, the oxalate dianion is protonated to produce the monoanion and oxalic acid, which cannot act as an electron donor in the photocatalytic system. At higher pH, reduction of O2 is thermodynamically unfavourable, because the one-electron reduced species of O2 (O2˙−) cannot be protonated to produce HO2˙ that disproportionates to yield H2O2 and O2. Thus, the H2O2 formation rate reached maximum around pH 4.5, which is close to the pKa2 of oxalic acid (4.27) and the pKa of HO2˙ (4.9).13
![(a) Time courses of H2O2 production by photoirradiation (λ > 340 nm) of an aqueous suspension containing (COO−)2 (400 mM, red; 200 mM, blue; 100 mM, black, [(COOK)2]/[(COOH)2] = 3) and the QuPh+–NA@sAlMCM-41 composite ([QuPh+–NA]: 0.2 mM). (b) Time courses of H2O2 production under different pH conditions. The pH value of each aqueous medium was controlled to 1.1 (black), 4.5 (red) or 5.5 (blue) by changing the ratio of (COOH)2 to (COOK)2. The total concentration of the oxalate anion was fixed at 200 mM.](/image/article/2013/CC/c3cc41575a/c3cc41575a-f3.gif) | ||
| Fig. 3 (a) Time courses of H2O2 production by photoirradiation (λ > 340 nm) of an aqueous suspension containing (COO−)2 (400 mM, red; 200 mM, blue; 100 mM, black, [(COOK)2]/[(COOH)2] = 3) and the QuPh+–NA@sAlMCM-41 composite ([QuPh+–NA]: 0.2 mM). (b) Time courses of H2O2 production under different pH conditions. The pH value of each aqueous medium was controlled to 1.1 (black), 4.5 (red) or 5.5 (blue) by changing the ratio of (COOH)2 to (COOK)2. The total concentration of the oxalate anion was fixed at 200 mM. | ||
The efficiency of the photocatalytic reaction can be evaluated by the quantum yield (Φ) of the products, where the Φ value is defined as the mole number of H2O2 produced divided by that of photons absorbed by the photocatalyst. In previous reports, the Φ value of H2O2 production has been determined to be 4.2% by photoirradiation (340 nm) of an oxygen-saturated buffer (pH 7.5–8.0) containing ZnO colloid and oxalate.14 The Φ value of the H2O2 production in an aqueous solution containing the QuPh+–NA@sAlMCM-41 composite ([QuPh+–NA]: 0.2 mM) and oxalate (200 mM) was determined to be 10% using a ferrioxalate actinometer under photoirradiation with monochromatised light of 334 nm. Such a high quantum yield originated from the high oxidizing and reducing abilities of photogenarated QuPh˙–NA˙+, which are sufficient for both the oxidation of oxalate and the reduction of O2.5
In summary, a long-lived ET state was attained in pure water for the first time by supporting QuPh+–NA on nanosized mesoporous silica–alumina. The formation of the long-lived ET state of the QuPh+–NA@sAlMCM-41 composite was confirmed by EPR measurements. The composite has been shown to act as an efficient photocatalyst for H2O2 production by reducing O2 with oxalate in water. The high quantum yield of 10% for H2O2 production in water without any organic solvent was comparable to that achieved in the homogeneous reaction system using an organic solvent.15
Notes and references
- (a) H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS; (b) D. G. Nocera, Chem. Soc. Rev., 2009, 38, 13 RSC; (c) J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802 CrossRef CAS; (d) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS; (e) S. Fukuzumi, Eur. J. Inorg. Chem., 2008, 1351 CrossRef CAS.
- (a) D. Gust, T. A. Moore and A. L. Moore, in From Non-Covalent Assemblies to Molecular Machines, ed. J. P. Sauvage and P. Gaspard, Wiley-VCH, Weinheim, 2011, pp. 321–354 Search PubMed; (b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS; (c) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283 RSC; (d) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS; (e) S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575 RSC.
- (a) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (b) S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427 RSC; (c) N. Martin, L. Sanchez, M. A. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015 CrossRef CAS; (d) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177 CrossRef CAS; (e) M. R. Wasielewski, J. Org. Chem., 2006, 71, 5051 CrossRef CAS.
- (a) H. Imahori, D. M. Guldi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 6617 CrossRef CAS; (b) D. M. Guldi, H. Imahori, K. Tamaki, Y. Kashiwagi, H. Yamada, S. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2004, 108, 541 CrossRef CAS; (c) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303 CrossRef CAS; (d) M. Murakami, K. Ohkubo, T. Nanjo, K. Souma, N. Suzuki and S. Fukuzumi, ChemPhysChem, 2010, 11, 2594 CrossRef CAS.
- (a) H. Kotani, K. Ohkubo and S. Fukuzumi, Faraday Discuss., 2011, 18, 89 Search PubMed; (b) Y. Yamada, T. Miyahigashi, H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 16136 CrossRef CAS; (c) Y. Yamada, T. Miyahigashi, H. Kotani, K. Ohkubo and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 6111 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS.
- S. Fukuzumi, K. Doi, A. Itoh, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15572 Search PubMed.
- A. Szegedi, Z. Konya, D. Mehn, E. Solymar, G. Pal-Borbely, Z. E. Horvarth, L. P. Biro and I. Kiricsi, Appl. Catal., A, 2004, 272, 257 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CAS.
- Standard Potentials in Aqueous Solution, ed. A. J. Bard, R. Parsons and J. Jordan, Marcel Dekker, New York, 1985 Search PubMed.
- D. B. Haytowitz and R. H. Matthews, Agriculture Handbook No. 8–11, Science and Education Administration, USDA, Washington, D. C., 1984 Search PubMed.
- A. Hodgkinson, Oxalic Acid in Biology and Medicine, Academic Press, London, 1977 Search PubMed.
- (a) R. C. Weast, Handbook of Chemistry and Physics 56th Edition, CRC, Cleaveland, OH, 1975–1976 Search PubMed; (b) B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14.
- A. J. Hoffman, E. R. Carraway and M. R. Hoffmann, Environ. Sci. Technol., 1994, 28, 776.
- Y. Yamada, A. Nomura, T. Miyahigashi and S. Fukuzumi, Chem. Commun., 2012, 48, 8329 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S4 (TEM images of Na+-exchanged sAlMCM-41, the N2 isotherm curve and the BJH plot, UV-vis absorption change and time courses of EPR signals). See DOI: 10.1039/c3cc41575a |
| This journal is © The Royal Society of Chemistry 2013 |