 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroscopic origin of the optical processes in blue sapphire†
Jessica K.
Bristow
a,
Stephen C.
Parker
a,
C. Richard
A. Catlow
b,
Scott M.
Woodley
b and
Aron
Walsh
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: a.walsh@bath.ac.uk
bUniversity College London, Kathleen Lonsdale Materials Chemistry, Gower Street, London, WC1E 6BT, UK
First published on 1st May 2013
Abstract
Al2O3 changes from transparent to a range of intense colours depending on the chemical impurities present. In blue sapphire, Fe and Ti are incorporated; however, the chemical process that gives rise to the colour has long been debated. Atomistic modelling identifies charge transfer from Ti(III) to Fe(III) as being responsible for the characteristic blue appearance.
Sapphire is a widely used industrial material for applications such as lasers, catalyst supports, and as substrate layers in photovoltaic devices.1–3 Pure corundum (α-Al2O3) is a ceramic, physically tough, transparent material. With the presence of iron and titanium chemical impurities, corundum crystals become vibrant blue sapphires. Isolated Fe(III) and Ti(III) incorporation results in yellow/green and pink crystals, respectively, so their chemical interaction is required to produce the blue response.
Previous work has given extensive theoretical insight into the properties of sapphire.4–7 These and many other studies have evaluated point defect behaviour and crystal imperfections, but have not directly connected these findings to the colour of the vibrant blue gem. The debate as to the chemical process, which is responsible for the colour of sapphire, has not yet been concluded. Experimental analysis, principally absorption spectroscopy, confirms there to be two possible optical mechanisms.8,9 The first, intervalence charge transfer (IVCT) between Ti(IV) and Fe(II) when situated in neighbouring lattice sites. The second, intra-atomic d–d transitions in one, or multiple, transition metal impurities. Previous analysis has been unable to distinguish which is responsible for its colour and what is the underlying chemical process.10,11
The first of the two possible mechanisms is an IVCT between neighbouring titanium and iron cations in the sapphire structure; the corresponding geometry is illustrated in Fig. 1. Impurity metals have been shown by such techniques as EDXRF (X-ray fluorescence) to directly substitute the host aluminium lattice sites. These cations remain in the same distorted octahedral D3d environment as Al, only expanded with the increased radii of the metal impurities.8 When in neighbouring lattice sites, charge transfer between particular cations such as iron and titanium can occur:
| Fe3+ + Ti3+ → Fe2+ + Ti4+ | (1) |
The electron transfer process results in the absorption of energy. It is the overlap of d-orbitals on these metals that facilitates the efficient transfer of the electron along the chains of the edge and face-sharing metal octahedra. No optical selection rules (Laporte or spin) limit this IVCT; therefore, the colour of a material, if governed by this high intensity optical process, would be vibrant. Concentrations of iron and titanium differ between samples depending on the environment that the crystals have formed under and the history of the sample. Typical concentrations are 750 and 1500 ppm for titanium and iron impurities, respectively, but these can range between 280–21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm in blue sapphires.8,12 Titanium and iron were first confirmed to be present on adjacent lattice sites in sapphire by Burns and Burns (1984) using Mössbauer spectrometry.13 It was suggested by Eigenmann and Günthard (1972) that these could be in either valence state (II–IV or III–III pairs).14
000 ppm in blue sapphires.8,12 Titanium and iron were first confirmed to be present on adjacent lattice sites in sapphire by Burns and Burns (1984) using Mössbauer spectrometry.13 It was suggested by Eigenmann and Günthard (1972) that these could be in either valence state (II–IV or III–III pairs).14
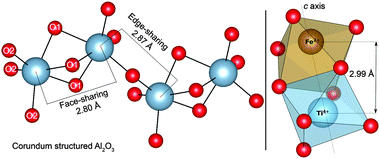 | ||
| Fig. 1 Illustration of the distorted Al-centred octahedra present in face-sharing and edge-sharing configurations in bulk corundum (left). Face-sharing nearest-neighbour titanium and iron oxygen octahedra are also drawn (right). | ||
In this Communication we investigate the origin of the optical processes in sapphire using an atomistic materials modelling approach. A polarisable interatomic potential, employed to model the charge transfer reactions, is combined with the angular overlap model to probe crystal field transitions.15 Separation of the electronic and ionic relaxation processes allows us to assess optical and thermal excitation energies independently. Full details of these methods are provided as ESI.†
A configurational coordinate diagram, depicting the relative energy change between two ionic configurations as a function of the local structure, has been calculated (Fig. 2). Our results (summarised in Table 1) show a vertical absorption at 670 nm for the 〈001〉 Fe–Ti pair, as governed by the Franck–Condon principle, to be present between the two ionic configurations. For the corresponding edge-sharing configuration, which is only 33 meV less stable than the face-sharing configuration, a charge transfer energy of 705 nm (1.76 eV) is calculated. This optical process will absorb energy close to the appropriate region of the electromagnetic spectrum for sapphires to be blue (the complementary colour of orange).
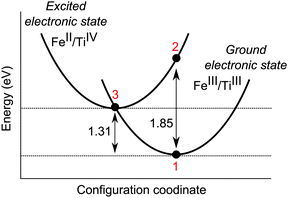 | ||
| Fig. 2 Configurational coordinate diagram for face-sharing iron and titanium in the corundum lattice. The vertical Franck–Condon transition (1 to 2) corresponds to 1.85 eV, while the thermal (zero phonon) transition (1 to 3) corresponds to 1.31 eV. | ||
| Chemical process (Eopt) | Energy (eV) | Energy (nm) |
|---|---|---|
| Fe3+ + Ti3+ → Fe2+ + Ti4+ | 1.85 | 670 |
The calculated energies are in good agreement with available experimental values as reported by Fontana et al. (2008) and Bonizzoni et al. (2009). Absorption spectroscopy identifies the characteristic broad, high intensity band with optical absorption perpendicular and parallel to the c axis reported as 571 nm (2.17 eV) and 693 nm (1.79 eV), respectively, to be that of the intervalence charge transfer.8,10 With the proven accuracy of these computational techniques, a comparison of these calculated energies is appropriate, and suggests these bands correspond to Eopt (Fe3+ + Ti3+ → Fe2+ + Ti4+). Additional interactions between multiple defects and the host lattice may account for the differences in the energy calculated with those of experiment. In particular, these calculations are formally athermal and neglect contributions from lattice vibrations.
Also shown in the configuration coordinate diagram (Fig. 2) is that the Fe(II)–Ti(IV) configuration is metastable with respect to electron transfer towards the trivalent configuration. It is the rate of this recombination process that will determine the lifetime of the metastable II–IV pairs in the sapphire structure, which are present as a consequence of photo-activation. Additional Fe(II) may also be formed, e.g., through interaction with lattice defects such as oxygen vacancies; however, in naturally occurring sapphires, the predominance of Fe(III) has been noted.16
Additionally, we have calculated whether the pairs of impurity ions are more stable as isolated species, or when situated in neighbouring lattice sites. Our results show a stabilisation of 0.64 eV when Fe(II) and Ti(IV) ions aggregate, due to their electrostatic attraction. The same method applied to the trivalent pair of cations reveals an energy stabilisation of 0.02 eV. The binding energy favours clustering of Fe and Ti, therefore enhancing the probability of IVCT occurring.
Recent studies by Achiwawanich et al. (2007) report that when heated in an inert atmosphere, the intensity of the dark blue colour of sapphire increases up to approximately 1500 K.19 This evidence seems to support our conclusion that IVCT is responsible for the colour of sapphire. The increased mobility of ions in the structure increases the probability of Fe–Ti pairs forming and hence charge transfer occurring. However, at very high temperatures configuration entropy will disfavour the aggregation of impurities and the excessive thermal vibrations may inhibit IVCT. Alternative, weaker optical processes would then dominate, with the loss of the vibrant blue colouration. The behaviour of doped-sapphire in a reducing atmosphere is more complex,17,18 and the role of oxygen related defects should also be considered.
The second possible mechanism as to why sapphire is blue is that internal crystal field transitions in impurity metals absorb light between 1.99–2.14 eV (624–580 nm); resulting in the characteristic colour. Although our calculations have confirmed that the intervalence charge transfer mechanism is likely to be responsible for the colour of sapphire, it has been argued that intra-valence transitions in metal impurities themselves are a contributing factor.9,10,20 It is for this reason that we have calculated the energies of these transitions. The metal impurities whose crystal field parameters we have investigated in sapphire are Cr3+, Ti3+, Fe3+, Fe2+, and Ni2+. Techniques in previous studies such as mass spectrometry and XRD identify these to be the chromophores that are found at the highest concentrations in the structure. Ti4+ has been omitted here as this cation is not optically active in the visible range and therefore cannot directly contribute to the colour of sapphire.11,19
The argument for the second mechanism, intra-valence d–d transitions in these ions, could also be supported by the experimental data on the effect of temperature on the colour of sapphire. Higher temperatures aid electron promotion through asymmetric thermal vibrations (breaking parity selection rules), thereby increasing the intensity of blue colouration.
Our results confirm that the substitutional transition metal centres (as has been widely reported) to be situated in anti-prismatic geometry (Fig. 1).11,21 Expansion in pairs of the oxide ligands during the formation of this geometry results in two Al–O bond lengths (see O1 and O2 in Fig. 1). We have calculated these bond lengths to be 1.98 Å and 1.84 Å. The corresponding Al–Al bond lengths are calculated to be 2.80 Å and 2.87 Å. This distortion from perfect octahedral geometry results in D3d symmetry, with the characteristic crystal field splitting diagram shown in Fig. 3. The intra-valence transitions for isolated impurities are listed in Table 2.
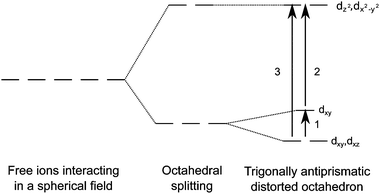 | ||
| Fig. 3 Splitting diagram of trigonal anti-prismatic geometry of metal centres in sapphire. | ||
| Cation | 1 | 2 | 3 | |
|---|---|---|---|---|
| (SF) – Spin forbidden | ||||
| Cr3+ | d3 | 0.16 (SF) | 1.31 | 1.47 |
| Ti3+ | d1 | 0.22 | 1.69 | 1.91 |
| Fe3+ | d5 | 0.18 (SF) | 0.97 (SF) | 1.15 (SF) |
| Fe2+ | d6 | 0.19 | 0.63 (SF) | 0.82 |
| Ni2+ | d8 | 0.08 | 0.29 | 0.37 |
Only isovalent cation impurities have crystal field transitions close to the energy range expected in order to be responsible for the colour of sapphire, which is in agreement with available experimental evidence. All optical transitions in isovalent Fe(III) are however spin-forbidden. Typical absorption intensities for these transitions are known to range from between 10−5–1 M−1 cm−1. These would therefore be too weak to be responsible for the intense colouration of the crystal.8
Corundum that has been only doped with an appropriate concentration of isovalent titanium has been shown to result in a pale pink coloured crystal, and is the basis for Ti sapphire lasers. Corundum that has been only doped with isovalent chromium forms vibrant red/pink rubies. Intra-atomic transitions in these chromophores have been shown to not absorb energy in the appropriate region of visible light to be responsible for the colour of blue sapphire.11,21,22
The calculated crystal field parameters correlate closely to those determined by absorption spectroscopy, and show the clear trends that would be expected based on the oxidation state of the central cation. These results validate the predicted analysis of the spectra for various samples of sapphire, and display an advantage of atomistic simulations in the separation and identification of individual signals in complex spectra.8,10
Our study has provided new insights into the optical properties of impurity cations commonly found in the structure of sapphire. Crystal field calculations have confirmed that intra-atomic transitions in impurity metals do not contribute to the colour of blue sapphire. We conclude, consistent with the available experimental evidence, that an intervalence charge transfer between Ti(III) and Fe(III) is the mechanism responsible for why sapphire is blue. Future work should consider both the interaction of impurities with point defects, as well as the interaction of impurities at the high-density limit, e.g. the formation of ilmenite (FeTiO3).
J.K.B. was funded by EPSRC (Grant No. EP/G03768X/1). S.C.P. would like to thank SRC and AERE Harwell for a CASE studentship. A.W. acknowledges support from the Royal Society University Research Fellowship scheme. We would like to thank A. A. Sokol and the late A. M. Stoneham for useful discussions. Access to the HECToR supercomputer was facilitated through membership of the HPC Materials Chemistry Consortium (Grant No. EP/F067496).
Notes and references
- E. F. Martynovich, V. I. Baryshnikov and V. A. Grigorov, Opt. Commun., 1985, 53, 257–258 CrossRef CAS.
- Y. J. Lee, M. H. Lee, C. M. Cheng and C. H. Yang, Appl. Phys. Lett., 2011, 98, 263504 CrossRef.
- K. F. Wall and A. Sanchez, Linc. Lab. J., 1990, 3, 447–462 Search PubMed.
- C. R. A. Catlow, R. James, W. C. Mackrodt and R. F. Stewart, Phys. Rev. B, 1982, 25, 1006 CrossRef CAS.
- P. W. M. Jacobs and E. A. Kotomin, Philos. Mag., 1993, 68, 695–709 CrossRef CAS.
- A. A. Sokol, A. Walsh and C. R. A. Catlow, Chem. Phys. Lett., 2010, 492, 44–48 CrossRef CAS.
- K. J. W. Atkinson, R. W. Grimes, M. R. Levy, Z. L. Coull and T. English, J. Eur. Ceram. Soc., 2003, 23, 3059–3070 CrossRef CAS.
- L. Bonizzoni, A. Galli, G. Spinolo and V. Palanza, Anal. Bioanal. Chem., 2009, 395, 2021–2027 CrossRef CAS.
- A. N. Platonov, A. N. Tarashchan, K. Langer, M. Andrut, G. Partzsch and S. S. Matsyuk, Phys. Chem. Miner., 1998, 25, 203–212 CrossRef CAS.
- I. Fontana, A. Le Donne, V. Palanza, S. Binetti and G. Spinolo, J. Phys.: Condens. Matter, 2008, 20, 012086 CrossRef.
- D. S. McClure, J. Chem. Phys., 1962, 36, 2757 CrossRef CAS.
- E. Gaudry, D. Cabaret, P. Sainctavit, C. Brouder, F. Mauri, J. Goulon and A. Rogalev, J. Phys.: Condens. Matter, 2005, 17, 5467–5480 CrossRef CAS.
- R. G. Burns and V. M. Burns, J. Adv. Ceram., 1984, 10, 46–61 CAS.
- K. Eigenmann and H. Günthard, Chem. Phys. Lett., 1972, 13, 58–61 CrossRef CAS.
- J. D. Gale, J. Chem. Soc., 1997, 93, 629–637 Search PubMed; C. R. A. Catlow, Proc. R. Soc. London, Ser. A, 1977, 353, 533–561 CrossRef CAS; S. M. Woodley, P. D. Battle, C. R. A. Catlow and J. D. Gale, J. Phys. Chem. B, 2001, 105, 6824–6830 CrossRef.
- V. Palanza, A. Galli, R. Lorenzi, F. Moretti, M. C. Mozzati, A. Paleari and G. Spinolo, Mater. Sci. Eng., 2010, 15, 012086 Search PubMed; K. S. Jheeta and D. C. Jain, Afr. Rev. Phys., 2007, 1, 460–465 Search PubMed.
- T. Kittiauchawal and P. Limsuwan, Kasetsart J. (Nat. Sci.), 2007, 41, 262–266 Search PubMed.
- N. Udomkan, P. Limsuwan, P. Winotai and S. Meejoo, Int. J. Mod. Phys. B, 2005, 19, 3273–3284 CrossRef CAS.
- S. Achiwawanich, B. D. James and J. Liesegang, Appl. Surf. Sci., 2007, 253, 6883–6891 CrossRef CAS.
- S. L. Reddy, K. N. M. Reddy, N. C. G. Reddy, R. L. Frost and T. Endo, Mol. Phys., 2009, 107, 1537–1539 CrossRef CAS.
- P. Lacovara, L. Esterowitz and M. Kokta, Int. J. Quantum Electron., 1985, 21, 1614–1618 CrossRef.
- L. E. Orgel, Nature, 1957, 179, 1348–1348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed computational methods. See DOI: 10.1039/c3cc41506f |
| This journal is © The Royal Society of Chemistry 2013 |