 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new photoclick reaction strategy: photo-induced catalysis of the thiol-Michael addition via a caged primary amine†
Weixian
Xi
a,
Matthias
Krieger
a,
Christopher J.
Kloxin
b and
Christopher N.
Bowman
*a
aDepartment of Chemical and Biological Engineering, University of Colorado, Boulder, Colorado 80309-0424, USA. E-mail: christopher.bowman@colorado.edu; Fax: +1-303-492-4341; Tel: +1-303-492-3247
bDepartment of Materials Science & Engineering and Department of Chemical & Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, Delaware 19716, USA
First published on 8th March 2013
Abstract
The utilization of 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) as a photolabile primary amine cage enables the thiol-Michael ‘click’ reaction to be photo-triggered. The photolabile amine exhibits efficient catalytic activity upon UV irradiation and is shown to initiate the photopolymerization of tetrathiol and diacrylate comonomers viaMichael addition.
The thiol-Michael addition is one of the most widely implemented ‘click’ reactions in polymer science, being utilized in dendrimer synthesis and polymer network formation as well as bioconjugation and polymer functionalization.1–3 The thiol-Michael reaction is readily catalyzed through either a base- and nucleophile-pathway;4,5 however, there are no light-mediated methods to control this reaction which would have a significant impact in several material fabrication strategies. Moreover, there are relatively few click reactions that are amendable to photoinitiation, such as the radical-mediated thiol–ene and photoreduced Cu-catalyzed azide–alkyne cycloaddition reactions.2,6–8 Some commercially available photogenerated acids/bases have been used in many reactions such as thiol-epoxide reaction.9 This lack of utilization is likely due to the fact that their photolysis mechanism often includes the generation of radicals which would lead to acrylate (or other vinyl monomer) homopolymerization, rendering it inappropriate for Michael addition. Therefore, there is a need and challenge for developing novel light controlled reactions in both polymer chemistry and material science to expand and diversify the photo-mediated click reaction toolbox. In this communication, we introduce the concept of utilizing a photolabile catalyst for initiation of the thiol-Michael addition reaction to achieve spatiotemporal control of this highly versatile and implemented coupling reaction.
The 2-nitrobenzyl functional group is perhaps the most common photolabile protecting group utilized in organic synthesis (e.g., OH, NH2, SH, COOH, etc.).10 In the last few decades, several 2-nitrobenzyl derivatives have been developed with better quantitative yields and photolytic efficiency. In particular, 2-nitroveratryloxycarbonyl (NVOC) and 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) have been used as primary amine protecting groups for spatioselective surface functionalization and photolithographic synthesis of oligo nucleopeptide/peptide microarray.11–17Primary amines, such as hexylamine, are efficient catalysts for the thiol-Michael addition in macromolecular synthesis, but lack the spatiotemporal control afforded by photoinitiation.5,18 In this study, we hypothesize that NPPOC will be an effective protecting group for hexylamine, which will be released upon UV irradiation to catalyze the thiol-Michael addition of a thiol and acrylate. We chose NPPOC as our photolabile functional group because of its high quantum yield during photolysis as compared with NVOC (Scheme 1).19
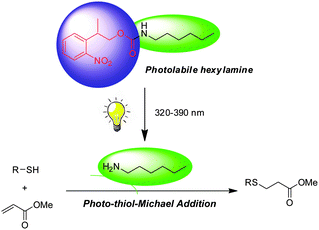 | ||
| Scheme 1 Photo-triggered thiol-Michael addition reaction using NPPOC-hexylamine as a catalyst. | ||
NPPOC-hexylamine was prepared by direct amidation and was subsequently photo-deprotected using 320–390 nm UV light (see ESI†). The photolytic deprotection reaction of NPPOC-hexylamine was monitored by UV/Vis (Fig. 1). This photolytic reaction proceeded rapidly, finishing in 30 min based on UV/Vis spectroscopy. Previous researchers have elucidated two identical pathways for NPPOC photo-cleavage (Fig. 1a): the nitroso pathway, which usually occurs with 2-nitrobenzyl-type protecting groups, and β-elimination, which occurs in the presence of water or an amine base.15,16,20 The UV/Vis spectra reveal the initial formation of the nitroso product, having typical peak locations at 290 and 317 nm (Fig. 1b). These peaks then decrease with increasing irradiation time, which is attributed to the suppression of the nitroso pathway by the newly released hexylamine, in accordance with previous work.16 Thus, in the photolytic reaction of NPPOC-hexylamine, the β-elimination is favoured, which is further evidenced by 13C-NMR (see ESI†).
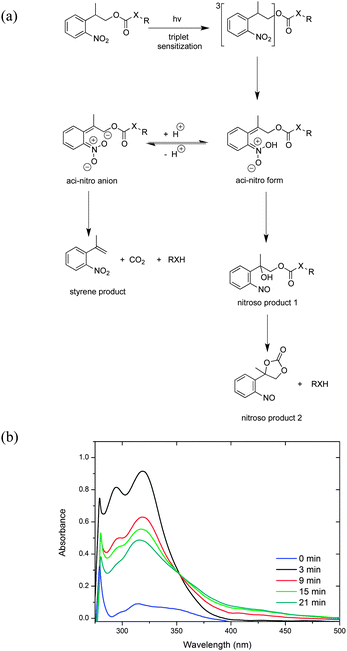 | ||
| Fig. 1 (a) Principle photo-cleavage pathways and products of the NPPOC-protected substrates. (b) UV/Vis spectra of NPPOC-hexylamine (0.05 mM) before and after photolysis using 320–390 nm irradiation in MeOH. | ||
To demonstrate the potential of NPPOC-hexylamine as a photocatalyst for the thiol-Michael addition reaction, we chose thiol glycolate and methyl acrylate as model substrates (Table 1). The model reaction gives over 90% yield in 1 h, indicating this catalytic reaction proceeds rapidly and efficiently. Control experiments (entries 6 and 7) indicate that both the photolabile catalyst and photo-irradiation are essential to trigger this reaction.
| Entry | R–SH | Catalyst loading (mol%) | Irradiation time (min) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: thiol (1 mmol), methyl acrylate (1 mmol) and NPPOC-hexylamine (0.05 mmol) under 320–390 nm irradiation (20 mW cm−2). b 1H-NMR yield. c Irradiation time is 0 min, reaction time is 120 min. | ||||
| 1 |

|
5 | 60 | 93 |
| 2 |

|
5 | 60 | 94 |
| 3 |

|
5 | 60 | 91 |
| 4 |

|
5 | 90 | 93 |
| 5 |
|
1 | 90 | 55 |
| 6 |

|
5 | 0c | 0 |
| 7 |
|
0 | 90 | Trace |
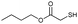
Real-time Fourier transform infrared (FTIR) spectroscopy reveals that the photo-induced catalysis of the thiol-Michael reaction proceeds in two stages (Fig. 2). The first stage is characterized by slow reaction kinetics, the duration of which exhibits a weak dependence on irradiation time. Notably, 2 and 4 minutes of irradiation show a similar, delayed upturn (∼6 min) in the conversion trajectory towards the second stage of rapid reaction kinetics. As these are thin film specimens, these trends are not attributable to any thermally induced autoacceleration effect. One possible explanation is that the conversion from the aci-nitro intermediate to the product is initially slow; however, upon the formation of the basic products (Fig. 1a), such as the newly released amine (RNH2), thiol anion (RS−), or carbon anion (RCH−COOMe), the deprotonatation of the aci-nitro intermediate pathway becomes favoured. As noted above, the β-elimination styrene product, which follows this reaction pathway, is observed in 13C-NMR (see ESI†). Presuming that the kinetics of this nucleophile-initiated pathway is more rapid, combined with the increased basicity of the solution, the sudden increase in kinetics shown in Fig. 2 is explained. Additionally, no reaction occurs in the absence of irradiation, which in accordance with our 1H-NMR results.
 | ||
| Fig. 2 Thiol conversion versus time, as measured by FTIR, for a stoichiometric mixture of butyl thiol glycolate and methyl acrylate using 5% NPPOC-hexylamine under continuous (filled square), 4 min (open triangle), 2 min 320–390 nm irradiation (filled triangle), and no irradiation (open circle) as well as no NPPOC-hexylamine under continuous irradiation (filled circle). | ||
Finally, we demonstrated the use of NPPOC-hexylamine as a photo-catalyst for the thiol-Michael photopolymerization reaction. We chose two tetrathiols (PETMA and PETMP) and a bisphenol A ethoxy diacrylate (BPAEDA) as substrates for the polymer network formation. To mitigate the side effects of CO2, which is released during photolysis, the NPPOC-hexylamine concentration was decreased from 5 to 3 mol% and the irradiation time was extended to 1 hour. While similar, the resulting polymer networks exhibited a slight shift in glass transition temperatures as determined using dynamic mechanical analysis (DMA) via the peak in the tan δ curve (i.e., 29 and 25 °C for the PETMA/BPAEDA, Fig. 3a, and PETMP/BPAEDA, Fig. 3b, respectively). The larger elastic moduli in the rubbery region for the PETMA- versus PETMP-based material suggests an increased crosslink density (0.95 M versus 0.77 M at 80 °C), which is attributed to the shorter distance between crosslinks in the PETMA system. In either case, the narrow tan δ peaks indicate a homogeneous polymer network as observed in other thiol–ene photopolymerization reactions.2,3
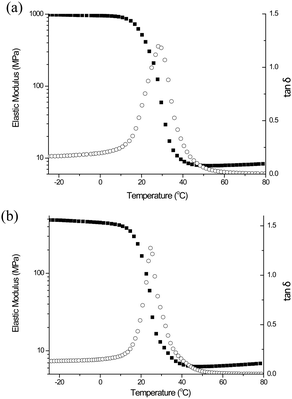 | ||
| Fig. 3 Tan δ and elastic modulus plots vs. temperature for networks formed from a stoichiometric mixture of PETMA (a), PETMP (b) and bisphenol A ethoxy diacrylate. | ||
In summary, we developed a photolabile, caged primary amine (NPPOC-hexylamine) as a photo-catalyst for the thiol-Michael addition reaction and polymerization. This catalyst has efficient catalytic activity upon 320–390 nm irradiation. We demonstrated the utility of photolabile caged amines in photo-initiated thiol-Michael polymer network formation with the associated advantages of a homogeneous network with a sharp, well-defined glass transition.
This work was supported by the National Scientific Foundation grant CHE-1214109.
Notes and references
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- W. X. Xi, C. Wang, C. J. Kloxin and C. N. Bowman, ACS Macro Lett., 2012, 1, 811–814 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- B. J. Adzima, Y. H. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS.
- S. C. Ritter and B. Konig, Chem. Commun., 2006, 4694–4696 RSC.
- J. W. Chan, H. Zhou, C. E. Hoyle and A. B. Lowe, Chem. Mater., 2009, 21, 1579–1585 CrossRef CAS.
- C. M. Seubert and M. E. Nichols, J. Coat. Technol. Res., 2010, 7, 615–622 CrossRef CAS.
- M. Zabadal and P. Klan, Chem. Listy, 2001, 95, 694–699 CAS.
- K. R. Bhushan, C. DeLisi and R. A. Laursen, Tetrahedron Lett., 2003, 44, 8585–8588 CrossRef CAS.
- H. Giegrich, S. Eisele-Buhler, C. Hermann, E. Kvasyuk, R. Charubala and W. Pfleiderer, Nucleosides Nucleotides, 1998, 17, 1987–1996 CAS.
- Z. C. Liu, D. S. Shin, K. T. Lee, B. H. Jun, Y. K. Kim and Y. S. Lee, Tetrahedron, 2005, 61, 7967–7973 CrossRef CAS.
- S. J. Min, K. D. Ahn and J. M. Kim, Bull. Korean Chem. Soc., 2005, 26, 1437–1439 CrossRef CAS.
- D. Woll, J. Smirnova, M. Galetskaya, T. Prykota, J. Buhler, K. P. Stengele, W. Pfleiderer and U. E. Steiner, Chem.–Eur. J., 2008, 14, 6490–6497 CrossRef.
- H. Yi, S. Maisonneuve and J. Xie, Org. Biomol. Chem., 2009, 7, 3847–3854 CAS.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- A. Hasan, K. P. Stengele, H. Giegrich, P. Cornwell, K. R. Isham, R. A. Sachleben, W. Pfleiderer and R. S. Foote, Tetrahedron, 1997, 53, 4247–4264 CrossRef CAS.
- S. Walbert, W. Pfleiderer and U. E. Steiner, Helv. Chim. Acta, 2001, 84, 1601–1611 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc41123k |
| This journal is © The Royal Society of Chemistry 2013 |