 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalization of organic semiconductor crystals via the Diels–Alder reaction†
Brittni A.
Qualizza
,
Srividya
Prasad
,
M. Paul
Chiarelli
and
Jacob W.
Ciszek
*
Department of Chemistry and Biochemistry, Loyola University Chicago, 1032 W. Sheridan Rd, Chicago, Illinois 60660, USA. E-mail: jciszek@luc.edu; Fax: +1 773-508-3806; Tel: +1 773-508-3107
First published on 26th March 2013
Abstract
A surface adlayer is generated on organic single crystals (tetracene and rubrene) using the site specific Diels–Alder reaction and a series of vapor phase dienophiles. X-ray photoelectron spectroscopy (XPS) confirms adsorption on the surfaces of tetracene and rubrene and mass spectrometry demonstrates the reaction’s applicability to a range of dienophiles.
The functionalization of surfaces with small adsorbates has allowed scientists and engineers to manipulate material properties1 including tribology,2–4 adhesion,5–8 biocompatibility,9–11 corrosion resistance,12 catalysis,13 electronic band structure,14and luminescence.15 For example, monolayers of n-alkanethiols have reduced the rate of oxidation on copper; at a monolayer thickness of 25 Å, oxidation decreases by two orders of magnitude.12 Furthermore, in nanoparticle systems, the nature of the terminus on an adsorbed monolayer determines its attraction to and uptake into cells.9,10 Using established chemistries on common substrates such as coinage metals (gold, silver, copper),16 silicon,17 silicon dioxide,18 platinum,19 and alumina,20 to name but a few,21 these highly developed systems even allow for incorporation of complex functions into materials.19,22,23
The diversified properties of these reacted materials raised our interest in the possibility of functionalizing the surfaces of organic semiconductors. In fact, many of the significant flaws of organic semiconducting materials, for example, their propensity to oxidize,24 degradation resulting from humidity,25 and the prevalence of trap states at the semiconductor/dielectric interface32,33 have been classically remedied in similar systems via monolayers.26,27 It is also possible to change the electronic properties of the materials (carrier density, carrier mobility, conductivity, field-effect mobility) in devices based on surface effects,28,29 presenting a compelling motivation for a means of surface selective functionalization. In fact, an earlier report by Calhoun et al. demonstrated that functionalizing the surface of tetracene and rubrene with organosilanes (trichloro- or triethoxy-) produced a thin (presumably oligomerized) film that increased the surface conductivity of the crystal.28 If such work could be extended to a highly defined and perhaps milder reaction, a wider variety of functional groups could be appended to an organic surface with greater control over density and location, and eventually lead to the manipulation of properties.
In this vein, we report the surface functionalization of two organic semiconductors, tetracene (1) and rubrene (2), via the Diels–Alder reaction as a step towards generating ordered coverage on an organic single crystal surface. The Diels–Alder reaction is site-specific, well studied, and can accommodate a wide range of adsorbates (3–7, Fig. 1), potentially allowing for the organization and diversity of adsorbates previously exclusive to thiolate monolayers. Additionally, our initial studies of tetracene and rubrene imply that this reaction should be general to the acene class of organic semiconductors which find a significant role in current electronics.30
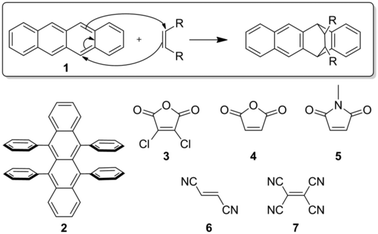 | ||
| Fig. 1 The Diels–Alder reaction occurring between the surface of single crystal acenes (the Diels–Alder dienes, 1, 2) and commercially available dienophiles (3–7). (top) The mechanism for the Diels–Alder reaction between tetracene and a generic dienophile. In the case of tetracene (1), the reaction preferentially occurs at the 5,12 position. (bottom left) In the case of rubrene (2), the reaction is predominately at the unsubstituted 1,4 position. | ||
Experiments were performed on single crystal surfaces. In contrast to thin-film organic materials, where various crystalline facets inhibit the study of well-defined chemical reactions, organic single crystals have very low surface defect densities and known surface molecular geometries.31,32 Single crystals of tetracene and rubrene were grown by physical-vapor transport in argon in a horizontal tube furnace33 and either collected off the furnace walls as free crystals or grown directly on slides. If crystals were removed more than an hour before analysis, they were stored in a nitrogen filled glovebox or a vacuum chamber in the dark. For a typical reaction, a single crystal was placed in a Schlenk round bottom flask equipped with a glass hollow stopper containing a dienophile and the crystal was placed approximately 8 cm away. The pressure in the flask was reduced (10−1 Torr) to remove residual volatiles and then heated to 85 °C (where all dienophiles display a vapor pressure of at least 0.27 Torr).34 Reactions were allowed to proceed for up to 3 days to ensure substantial coverage. Reacted substrates remain crystalline as determined by X-ray diffraction.
Two methods were employed to confirm adsorption of the dienophile on the surface, its chemical identity, and any potential face selectivity of the reaction: mass spectrometry and X-ray photoelectron spectroscopy (XPS). If necessary, these solid–vapor Diels–Alder reactions were compared to solution generated equivalents (sealed tube, toluene, 0.1–0.3 M, 120–150 °C, 8–72 h, see ESI†) so that the known adduct, once characterized, could be used as a standard. Such samples are referred to as the standard adduct throughout the paper.
To begin, we probed a reacted tetracene crystal surface for the presence of dienophile adsorbates. In order to provide a diagnostic XPS signal, which was not obscured by the elements present on a pristine surface, 2,3-dichloromaleic anhydride (3) was chosen as the dienophile. The lack of Cl 2p signal for an unreacted crystal can be seen in Fig. 2 (dotted line). In the reacted crystal, the characteristic Cl 2p3/2 and 2p1/2 signals are clearly seen. Importantly, the Cl 2p signal in Fig. 2 (solid line) (201.3, 202.9 eV) has a similar binding energy to that of the standard adduct (Fig. S1, ESI†) which was analyzed as a powder. Indirectly, these same data can also be used as an indication of chemisorption of the species: the low pressure (∼10−8 Torr) and ambient temperature of the XPS chamber suggest against physisorption in the case of our sample.35,36 Similar results were observed for rubrene, though the data here are suggestive of two distinct adsorbed species (Fig. S2, ESI†)
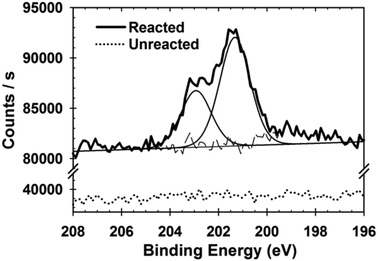 | ||
| Fig. 2 XPS studies of the Cl 2p core electrons for tetracene single crystals grown directly on gold slides demonstrating adsorption of 2,3-dichloromaleic anhydride (black line). Unreacted single crystals grown directly on gold slides (dotted line) demonstrated no Cl 2p signal. Signal binding energies were referenced to the C 1s signal occurring at 284.5 eV. | ||
To confirm the (chemisorbed) nature of the adsorbed species, mass spectrometry was used to identify the maleic anhydride adduct at the crystal surface. Here, a crystal exposed to maleic anhydride (4) was directly vaporized at 10−8 Torr via a thermal probe. Molecular ions and fragmentation patterns for the single crystal are identical to those of the standard adduct (Fig. S3, ESI†). Thermal experiments also allow us to conclude that the majority species is chemisorbed (Fig. S4, ESI†) based on a small but observable signal for physisorbed material, distinct from the prominent adduct signal. With chemical adsorption of the dienophile established, MS was also used to screen several tetracene–dienophile reactions for adduct formation; it was used to confirm the adducts of tetracene single crystals and N-methylmaleimide (5), fumarodinitrile (6), and tetracyanoethylene (7) (Fig. S5–S7, ESI†).
The previous mass spectrometry results also allow us to comment on the potential applicability of the Diels–Alder reaction to other reactants. Dienophiles (3–7) were chosen in order to cover a range of reactivity from highly activated olefins (7), to intermediate (3, 4, 5), to mild (6).37 The presence of an adduct for fumarodinitrile (6) is worth noting due to its low activity. We assume that a successful reaction is a consequence, in part, of the tetracene; a rather reactive diene. This acene core displays reaction rates with maleic anhydride more than 10 times as much as 1,3 butadiene, a standard diene. Other typical acene organic semiconductors (pentacene, rubrene) have been shown to be even more active toward dienophiles.38 This activity is important as we expect that the reaction must occur in a sterically demanding environment.
Theoretically, the fixed orientation of the reactive diene moiety within the single crystal could induce confinement effects,39 limiting the kinetics and possibly the thermodynamic stability of the system. Such effects should be predictable based on the crystallographic density of the acene core and its orientation with respect to the exposed surface and its nearest neighbors (Fig. 3). Crystallographic data of tetracene40 show that the largest face, the ab face (Fig. 3b), is primarily composed of a staggered herringbone structure with a nearest adjacent molecule approximately 5 Å apart. Tetracene molecules are in the same plane and the hydrogen on the adjacent molecules is approximately 3 Å distant from the neighboring acene core, presumably impeding the reaction. Instead, the other faces of the crystal, including the bc and ac, are expected to react with greater ease, based on a more exposed acene core (Fig. 3).
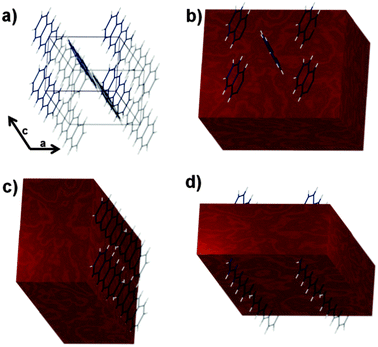 | ||
| Fig. 3 The crystal packing of tetracene and its arrangement relative to exposed crystal faces. (a) A simple unit cell for tetracene including crystallographic axes. Bond distances and molecular orientation can be found in ref. 40. (b–d) The unit cell shown with an overlaid macroscopic crystal face highlighting the portions of the molecules associated with the (ab) (ac) and (bc) faces of the unit cell. | ||
We observe evidence of the predicted differences in reactivity when examining single crystal tetracene crystals which have been reacted with 2,3-dicholoromaleic anhydride (2). Fig. 4a shows an optical microscopy image of a single crystal of tetracene with the ab face in the plane of the image. As can be seen in the Cl 2p region of the XPS spectrum (inset) no discernible chlorine signal can be seen. When a crystal is examined on its side (where a combination of other faces exist) a significant chlorine signal is observed. While this does not rule out reaction on the ab face, it does suggest that the rates for the various faces are different by orders of magnitude, much higher than reports of Diels–Alder reactions at sites with limited accessibility.41 It also indicates the potential for face selective reactions. Further study of this phenomenon is clearly warranted.
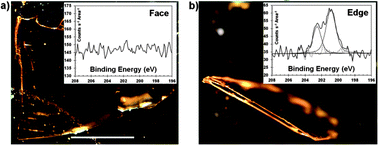 | ||
| Fig. 4 Optical images of the tetracene single crystals and their corresponding Cl 2p XPS spectra as insets. (a) The large face of a tetracene single crystal (scale bar is 1 mm) displays little/no reaction as determined by the lack of chlorine signal. (b) A single crystal placed on its side to expose the edge shows the presence of chlorine. Binding energies were referenced to the C 1s signal occurring at 284.5 eV and signals have been normalized to the area of the sample within the focal point of the beam. | ||
In summary, we have demonstrated that it is possible to dose adsorbates onto single crystals of tetracene and rubrene using the Diels–Alder reaction. A wide range of adsorbates containing functional groups such as imides, anhydrides, and cyano groups were applied, demonstrating this chemistry as a means to functionalize crystals of the acene class. While much exploration remains (e.g. rates of reaction and explicit surface coverage), potential exists for this chemistry to generate a defined, regular, and reproducible surface chemistry. Understanding these further properties and harnessing the capability to tailor the adlayer will surely lead to stimulating advancements in the theoretical and the experimental domain of organic materials, particularly at their surfaces.
JWC thanks NSF CAREER award #1056400 for supporting this work, Prof. Vitaly Podzorov for providing horizontal tube furnace schematics and advice on crystal growth, and Phillip Romanello for his assistance in furnace construction.
Notes and references
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS.
- R. Maboudian, W. R. Ashurst and C. Carraro, Tribol. Lett., 2002, 12, 95 CrossRef.
- J. W. Judy, Smart Mater. Struct., 2001, 10, 1115 CrossRef.
- T. M. Mayer, J. W. Elam, S. M. George, P. G. Kotula and R. S. Goeke, Appl. Phys. Lett., 2003, 82, 2883 CrossRef CAS.
- S. C. Clear and P. F. Nealy, J. Colloid Interface Sci., 1999, 213, 238–250 CrossRef CAS.
- S. R. Wasserman, H. Biebuyck and G. M. Whitesides, J. Mater. Res., 1989, 4, 886 CrossRef CAS.
- V. F. Petrenko and S. Peng, Can. J. Phys., 2003, 81, 387 CrossRef CAS.
- M. Mrksich, Chem. Soc. Rev., 2000, 29, 267 RSC.
- C. K. Kim, P. Ghosh, C. Pagliuca, Z.-J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818 CrossRef CAS.
- E. Ostuni, L. Yan and G. M. Whitesides, Colloids Surf., B, 1999, 15, 3 CrossRef CAS.
- P. E. Laibinis and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 9022 CrossRef CAS.
- Y. Uozumi and M. Nakazono, Adv. Synth. Catal., 2002, 344, 274 CrossRef CAS.
- M. Suda, N. Kameyama, A. Ikegami and Y. Einaga, J. Am. Chem. Soc., 2009, 131, 865 CrossRef CAS.
- M. Shim, C. Wang and P. Guyot-Sionnest, J. Phys. Chem. B, 2001, 105, 2369 CrossRef CAS.
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y.-T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152 CrossRef CAS.
- J. M. Buriak, Chem. Rev., 2002, 102, 1271 CrossRef CAS.
- J. Sagiv, J. Am. Chem. Soc., 1980, 102, 92 CrossRef CAS.
- J. W. Ciszek and J. M. Tour, Chem. Mater., 2005, 17, 5684 CrossRef CAS.
- T. W. Kelley, L. D. Boardman, T. D. Dunbar, D. V. Muyres, M. J. Pellerite and T. P. Smith, J. Phys. Chem. B, 2003, 107, 5877 CrossRef CAS.
- G. L. Hornyak, J. Dutta, H. F. Tibbals and A. K. Rao, Introduction to Nanoscience, CRC Press Boca Raton, 2008 Search PubMed.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704 CrossRef.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553 CrossRef CAS.
- D. Käfer and G. Witte, Phys. Chem. Chem. Phys., 2005, 7, 2850–2853 RSC.
- B. Park, Y. J. Kim, S. Graham and E. Reichmanis, ACS Appl. Mater. Interfaces, 2011, 3, 3545 CAS.
- L. Miozzo, A. Yassar and G. Horowitz, J. Mater. Chem., 2010, 20, 2513 RSC.
- J. Takeya, M. Yamagishi, Y. Tominari, R. Hirahara, Y. Nakazawa, T. Nishikawa, T. Kawaswe, T. Shimada and S. Ogawa, Appl. Phys. Lett., 2007, 90, 102120 CrossRef.
- M. F. Calhoun, J. Sanchez, D. Olaya, M. E. Gershenson and V. Podzorov, Nat. Mater., 2008, 7, 84 CrossRef CAS.
- V. Podzorov, S. E. Sysoev, E. Loginova, V. M. Pudalov and M. E. Gershenson, Appl. Phys. Lett., 2003, 83, 3504 CrossRef CAS.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS.
- R. M. Overney, L. Howald, J. Frommer, E. Meyer and H. J. Güntherodt, J. Chem. Phys., 1991, 91, 8441 CrossRef.
- E. Menard, A. Marchenko, V. Podzorov, M. E. Gershenson, D. Fichou and J. A. Rogers, Adv. Mater., 2006, 18, 1552 CrossRef CAS.
- R. A. Laudise, C. Kloc, P. G. Simpkins and T. Siegrist, J. Cryst. Growth, 1998, 187, 449 CrossRef CAS.
- R. H. Boyd, J. Chem. Phys., 1963, 38, 2529 CrossRef CAS.
- H. Koschel, G. Held, P. Trischberger, W. Widdra and H.-P. Steinrück, Surf. Sci., 1999, 437, 125 CrossRef CAS.
- J. Breitbach, D. Franke, G. Hamm, C. Becker and K. Wandelt, Surf. Sci., 2002, 507–510, 18 CrossRef CAS.
- V. D. Kiselev and A. I. Konovalov, J. Phys. Org. Chem., 2009, 22, 466 CrossRef CAS.
- D. Biermann and W. Schmidt, J. Am. Chem. Soc., 1980, 102, 3163 CrossRef CAS.
- I. Katash, X. Luo and C. N. Sukenik, Langmuir, 2008, 24, 10910 CrossRef CAS.
- D. Holmes, S. Kumaraswamy, A. J. Matzger and K. P. C. Vollhardt, Chem.–Eur. J, 1999, 5, 3399 CrossRef CAS.
- Y. Kwon and M. Mrksich, J. Am. Chem. Soc., 2002, 124, 806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for substrate preparation/single crystal growth; sample reaction conditions; XPS data for a standard tetracene adduct and reacted rubrene crystals; crystal mass spectrometry data; general synthetic experimental details for standard adducts; and 1H, 13C NMR, and IR characterization spectra. See DOI: 10.1039/c3cc40866c |
| This journal is © The Royal Society of Chemistry 2013 |